基质分散固相萃取-超高效液相色谱-串联高分辨质谱法同时测定干血斑样品中22种镇定催眠类药物
来源:优秀文章 发布时间:2023-02-17 点击:
于 峰, 魏 萌, 文 迪, 苗鑫刚, 张路迪, 张晓光
(1.河北医科大学法医学院,河北省法医学重点实验室,河北省法医分子鉴定协同创新中心,河北石家庄 050017;
2.河北医科大学护理学院,河北石家庄 050031;
3.河北医科大学法医鉴定中心,河北石家庄 050017;
4.河北医科大学,医学与健康研究院大型科研仪器设备共享服务平台,河北石家庄 050017)
目前较为广泛应用的镇定催眠类药物主要有苯二氮卓类、吩噻嗪类、巴比妥类等,这些药物具有改善睡眠、镇定、抵抗抑郁和焦虑等作用,但容易使患者产生依赖或者成瘾,过量服用会造成物理和精神的伤害,甚至呼吸衰竭死亡[1 -2 ]。因此,建立检测这些药物的准确快捷的方法十分必要[3]。干血斑法是指将采集的微量血样本置于采血卡上,经干燥后制成干血斑样品,其具有创伤小,便于储存和运输,后处理简单方便等优点。随着质谱技术在精准医学方面研究的开展,干血斑技术目前已经在疾病标志物筛查、药物动力学研究、临床药物监测等方面[4 -2 ]得到广泛的应用。
基质分散固相萃取法最初由美国路易斯安纳州立大学Staren Barker教授提出,它的原理是将硅胶键合有机相的净化填料放入萃取溶剂中,被检测物质和净化填料在剪切力作用下分散开来,被检测物质或者基质中杂质被吸附,使用不同极性溶剂进行解吸,从而达到净化目的[13 -15 ]。
目前,文献报道的检测镇定安眠药物的方法主要有气相色谱法、气相色谱-质谱联用法、液相色谱法、液相色谱-串联三重四级杆质谱法以及高分辨率质谱法等[16 -21 ]。静电场轨道阱高分辨率质谱是近年来兴起的先进的质谱分析技术,具有高通量、精确度高、分辨率高等优点,其通过记录被测物质加和离子在电场内的运动轨迹和频率,经过傅里叶转换得到精确质量数。使用干血斑结合静电场轨道阱高分辨率质谱法检测镇定催眠药物鲜有报道,与已有研究相比,本研究优势在于被测物种类全面,检出限低,分析时间短,前处理简单高效,通过对被分析物的精确质量数进行定性和定量,从而极大降低假阳性出现。
1.1 仪器、试剂与材料
QE-plus型超高效液相色谱-四极杆-静电场轨道阱高分辨率质谱仪(美国赛默飞世尔公司);
CT15RE型高速冷冻离心机(日本日立公司);
Vortex3000型涡旋混匀器(德国维根技术有限公司);
KQ-500E型超声波清洗器(昆山市超声仪器有限公司);
Waters Acquity BEH C18色谱柱(2.1×100 mm,1.7 μm,美国沃特世公司);
Thermo Scientific Hypersil GOLD C18色谱柱(2.1×100 mm,1.9 μm,美国赛默飞世尔科技公司);
Phenomenex Kinetex C18色谱柱(2.1×100 mm,2.6 μm,美国菲诺美公司);
Milli-Q Advantage A10超纯水仪(美国默克密理博公司) 。
22种安眠镇定类药物标准品(苯二氮卓类:奥沙西泮、地西泮、劳拉西泮、氯硝西泮、替马西泮、硝西泮、去甲西泮、7氨基氯硝西泮、氟西泮、氯氮平、阿普唑仑、艾司唑仑、咪达唑仑、三唑仑;
吩噻嗪类:丙咪嗪、氯丙嗪、异丙嗪、甲哌氯丙嗪;
巴比妥类:巴比妥、苯巴比妥、司可巴比妥、异戊巴比妥)浓度均为1 000 μg/mL,溶剂甲醇(坛墨质检-标准物质中心),3种内标标准品(地西泮D5、苯巴比妥D5、氯丙嗪D6)浓度均为100 μg/mL,溶剂甲醇(天津阿尔塔科技有限公司);
甲醇(色谱纯)、乙腈(色谱纯)、甲酸(99%)、乙酸铵(99%)、甲酸铵(99%)均购于美国默克密理博公司;
C18粉末(50 μm,60 Å)、PSA粉末(40~63 μm,60 Å)购于天津博纳艾杰尔公司;
Whatman 903采血卡(美国思拓凡公司);
实验用水为自制超纯水。
全血样品(河北医科大学法医鉴定中心提供)。
1.2 标准溶液配制
分别移取22种安眠镇定类药物标准品100 μL,用甲醇定溶于10 mL容量瓶中,配制成10 000 ng/mL的混标使用液1;
混标使用液1以甲醇稀释定容可分别制得1 000 ng/mL的混标使用液2及100 ng/mL的混标使用液3;
分别移取3种内标标准品1 000 μL,用甲醇定溶于10 mL容量瓶中,配制成10 000 ng/mL的混标内标使用液。
1.3 干血斑的制备
移取全血50 μL于Whatman 903采血卡上,温室条件下放置2 h得干血斑样品,置于-20 ℃冰箱保存。
1.4 样品前处理
沿虚线边缘处将干血斑裁剪下来,将其放入2 mL离心管中,加入10 μL(100 ng)混合内标使用液,40 μL甲醇水溶液(体积比1∶1)浸润干血斑,再加入950 μL乙腈,涡旋60 s,30 ℃超声5 min,10 000 g/min,4 ℃下离心5 min;
移取全部上清液于另一2 mL离心管中,加入PSA 100 mg,震荡混匀,涡旋60 s,10 000 g/min,4 ℃下离心5 min;
取上清液500 μL于另一2 mL离心管中,50 ℃水浴下氮气吹干,加入100 μL甲醇水溶液(体积比1∶3)复溶,10 000 g/min,4 ℃下离心1 min,取上清液上机测定。
1.5 基质加标标准溶液的制备
移取适量“1.2”混标使用液于9个2 mL离心管中,使离心管含有被测物质量依次为2 ng、5 ng、8 ng、20 ng、40 ng、80 ng、200 ng、400 ng、800 ng;
氮气小心吹干离心管,加入50 μL甲醇水溶液(体积比1∶1)复溶,涡旋30 s,再加入950 μL全血,涡旋混匀60 s,得到被测物浓度为2 ng/mL、5 ng/mL、8 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL、200 ng/mL、400 ng/mL、800 ng/mL的基质标准溶液。取制成的血液基质标准溶液各50 μL于Whatman 903采血卡上,温室条件下放置2 h即得,放置于-20 ℃冰箱保存。
1.6 基质效应评价方法
按照“1.3”制备干血斑,沿虚线边缘处将干血斑裁剪下来,将其放入2 mL离心管中,加入50 μL甲醇水溶液(体积比1∶1)浸润干血斑,加入950 μL乙腈,涡旋60 s,30 ℃超声5 min,10 000 g/min,4 ℃下离心5 min;
取上清液500 μL于另一2mL离心管中,50 ℃水浴下氮气吹干,移取“1.2”制备的适量标准溶液,加入甲醇水溶液(体积比1∶3)补足,使苯二氮卓类、吩噻嗪类添加浓度为10 ng/mL、400 ng/mL、600 ng/mL,巴比妥类添加浓度为40 ng/mL、400 ng/mL、600 ng/mL。同时用甲醇水溶液(体积比1∶3)直接配制成与上述浓度相同的标准溶液,上机测定,每个浓度水平重复测定3次。
1.7 外标法回收率测定方法
移取全血50 μL于Whatman903采血卡上(其中被测物浓度为40 ng/mL,400 ng/mL,600 ng/mL,按照“1.2”、“1.3”制备)不加入内标,改变PSA和C18加入量,加入PSA量依次为50 mg、100 mg、150 mg,加入C18量依次为50 mg、100 mg、150 mg,按照“1.4”进行样品前处理。
1.8 仪器条件
1.8.1 色谱条件色谱柱:Waters Acquity BEH C18色谱柱(100 mm×2.1 mm,1.7 μm)以及预柱(5 mm×2.1 mm,1.7 μm);
流动相相A 为10 mmol/L乙酸铵水溶液,B为乙腈溶液;
洗脱梯度:0~2 min,25%B;
2~9 min,25%B线性升高至90%B;
9~10 min,90%B;
10~11 min,90%B线性降低至25%B;
11~12 min,25%B;
流速0.3 mL/min,柱温30 ℃;
进样体积:2 μL。
1.8.2 质谱条件离子源:电喷雾电离源(HESI),离子源电压:3 800 V,离子源温度:350 ℃;
鞘气流速:40 arb;
辅助气流速:10 arb;
毛细传输管温度:320 ℃;
S-lens RF level:60;
扫描方式:一级质谱全扫描加数据依赖的二级质谱扫描(Full MS ddMS2);
扫描模式:正负切换;
一级质谱全扫描分辨率:70 000;
扫描范围:150~1 000m/z;
二级质谱扫描分辨率:17 500;
能量值(NCE stepped):20、30、40;
AGC target:1e5。
2.1 基质分散固相萃取条件的确定
2.1.1 基质效应评价按照“1.6”进行基质效应评价。基质加标标准溶液峰面积记为S,直接配制的相应浓度标准溶液峰面积记为S0,以(S-S0)/S0×100%评价基质效应。实验结果显示,实验中22种目标物质的基质效应范围为-20.7%至-8.5%之间,均为抑制效应。
2.1.2 净化剂的选择及用量实验选用C18和PSA作为净化剂,以回收率作为考察方式,确定净化剂的用量。内标和外标峰面积由于基质效应同时增强或者抑制,使用内标法计算回收率无法准确反映基质效应造成的影响,因此使用添加和不添加净化剂时的峰面积之比来考察净化剂的效果。
改变C18粉末和PSA粉末的用量,按照“1.7”进行实验,在仅添加C18的实验条件下,发现被测物回收率和不添加C18时峰面积无显著差异。在添加PSA粉末的情况下,除了司可巴比妥以外,其他被测物峰面积与不添加PSA粉末时相比均有不同程度的增加,这说明PSA对于血液样品具有更好的净化效果。其原因可能是PSA的氨基可与金属离子产生鳌合作用,从而有效的吸附了血液提取液中的金属离子以及脂类、有机酸、糖类,降低了基质效应。具体峰面积变化和添加量之间的关系如图1所示(被测物浓度为40 ng/mL),但是随着添加量的增多,硝西泮、地西泮、异丙嗪、司可巴比妥、巴比妥等峰面积出现了明显下降,这可能是由于PSA对被测物的吸附作用增强导致,在被测物浓度为400 ng/mL以及600 ng/mL的情况下改变PSA用量,也有相似的趋势。综合考虑,PSA添加量确定为100 mg。

图1 添加不同量的PSA净化剂条件下峰面积变化情况Fig.1 Effect of different dosage PSA on peak area a.benzodiazepines;b.phenothiazines;c.barbiturates.
2.2 色谱条件的优化
2.2.1 色谱柱的选择比较了Waters Acquity BEH C18色谱柱(100 mm×2.1 mm,1.7 μm),Thermo Scientific Hypersil GOLD C18色谱柱(100 mm×2.1 mm,1.9 μm)和Phenomenex Kinetex C18色谱柱(100 mm×2.1 mm,2.6 μm)出峰和分离情况。结果显示,3种色谱柱在12 min内均可以实现22种被分析物质的色谱分离。但是Hypersil GOLD C18色谱柱对于苯二氮卓类的分离效果和其他两种色谱柱相比出峰时间更加集中,Kinetex C18色谱柱色谱柱对于巴比妥类的分离效果和其他两种色谱柱相比出峰时间更加集中;
Acquity BEH C18色谱柱在分离效果方面表现得更加均衡。在Waters Acquity BEH C18色谱柱上吩噻嗪类和巴比妥类出峰峰形较其余两种色谱柱更加对称和尖锐,最终选择Waters Acquity BEH C18色谱柱作为分析柱。22种被测物的离子流色谱图见图2。

图2 22种被测物离子流色谱图Fig.2 Extracted ion chromatogram of 22 analytes
2.2.2 流动相的选择比较了甲酸铵水溶液-甲醇、甲酸铵水溶液-乙腈、乙酸铵水溶液-甲醇、乙酸铵水溶液-乙腈4种不同的流动体系下,22种被测物的出峰和分离情况。结果显示,使用乙腈作为有机相时,被测物出峰时间要早于使用甲醇作为有机相的出峰时间。对于地西泮、氯丙嗪、异丙嗪、甲哌异丙嗪、丙咪嗪,使用甲醇出峰时间均在8~10 min,此时有机相的比例最高(90%),共流出杂质增多,基质效应增强,导致出峰面积下降;
此外,对于巴比妥类物质,使用乙腈作为流动相时峰面积要明显大于使用甲醇作为流动相的峰面积。使用甲酸铵水溶液作为流动相和乙酸铵水溶液相比,苯二氮卓类和吩噻嗪类在峰面积、峰形方面无明显差异,但是对于巴比妥类物质,使用乙酸铵水溶液要大于使用甲酸铵水溶液出峰峰面积,因此最终选择使用乙酸铵水溶液-乙腈体系作为流动相。
2.3 质谱条件的优化
由于苯二氮卓类和吩噻嗪类物质离子化为[M+H]正模式,而巴比妥类离子化为[M-H]负模式,本仪器可以实现正负切换扫描,理论上可以节省一半仪器运行时间,但是正负切换扫描可能会带来扫描信息的丢失,因此本实验比较了正负切换同时扫描、正模式扫描、负模式扫描3种扫描方式下22种被测物质的出峰情况。结果表明,苯二氮卓类和吩噻嗪类正负切换扫描下灵敏度、峰面积和单正模式扫描下无明显差异,巴比妥类在正负切换扫描下峰面积出现了一定下降,但是下降幅度在10%之内,在正负切换扫描模式下,22种被测物灵敏度、线性范围、稳定性表现良好。综合考虑,本方法最终确定使用正负切换扫描,以提取的母离子精确质量数定性和定量,以提取的子离子精确质量数作为辅助定性,22种被测物的仪器实际提取质量数与理论质量数误差均在5 ppm之内,极大的减少了传统四极杆质谱假阳性或者假阴性的出现。22种被测物的分子式,理论质量数,典型子离子见表1。
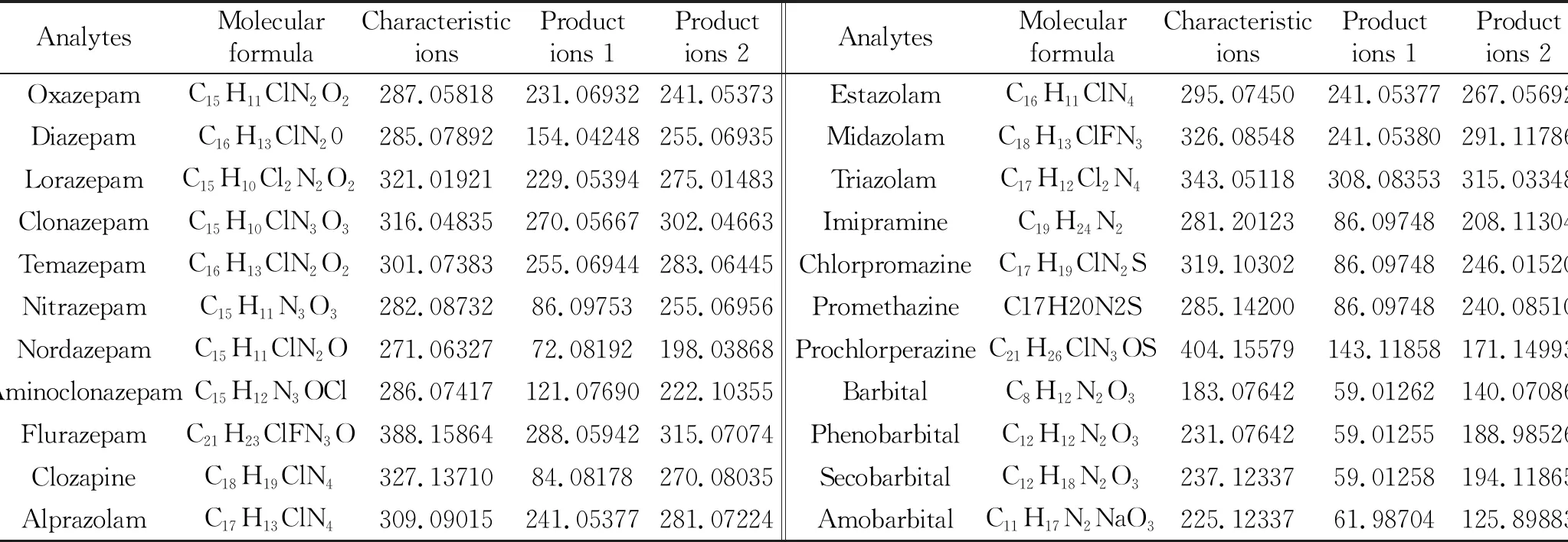
表1 22种被测物分子式、特征离子(n=5)
2.4 方法学验证
2.4.1 检出限、定量限、线性方程和线性范围按照“1.5”制作干血斑样品标准曲线,以外标峰面积和内标峰面积比值作为纵坐标y(其中苯二氮卓类以地西泮D5为内标、吩噻嗪类以氯丙嗪D6为内标、巴比妥类以苯巴比妥D5为内标),以被测物浓度作为横坐标x,进行线性拟合,得到线性方程。以3倍信噪比和10倍信噪比确定方法的检出限(LOD)和定量限(LOQ),结果如表2所示。苯二氮卓类、吩噻嗪类在5 ng/mL~800 ng/mL范围内线性表现良好(r2>0.99),巴比妥类在20 ng/mL~800 ng/mL范围内线性表现良好(r2>0.99),满足了日常司法检测的需要。
2.4.2 回收率和精密度取空白血样,添加低、中、高三水平的被测物(苯二氮卓类、吩噻嗪类添加浓度为低水平10 ng/mL、中水平400 ng/mL、高水平600 ng/mL,巴比妥类添加浓度为低水平40 ng/mL、中水平400 ng/mL、高水平600 ng/mL),按照1.2、1.3、1.4进行前处理,每个水平重复测定5次,计算平均回收率和相对标准偏差(RSD)见下表2。结果表明,22种被测物回收率在80.2%~113.5%范围内,RSD在0.9%~14.3%范围内,满足方法学要求。

表2 22种被测物线性方程、检出限、定量限、回收率和相对标准偏差(n=5)
2.4.3 样品稳定性取空白血样,添加低、中、高3水平(苯二氮卓类、吩噻嗪类添加浓度为10 ng/mL、400 ng/mL、600 ng/mL,巴比妥类添加浓度为40 ng/mL、400 ng/mL、600 ng/mL)的被测物,按照“1.2”、“1.3”制备干血斑样品,分成3组,分储于4 ℃、-20 ℃、-40 ℃条件下,在1、2、3、5、7、10、15、20、30天后进行测定,每组每个水平平行测定5次。结果表明,在-20 ℃以及-40 ℃保存条件下,被测物质30天内稳定性良好,测定结果RSD值在15%之内。4 ℃下,被测物7内稳定性良好,但是7天后稳定性变差,地西泮、地西泮、劳拉西泮、替马西泮、硝西泮、异丙嗪、苯巴比妥测定结果RSD值高于20%。
2.5 实际样品测定
收集了3份非正常死亡人员阳性血样,应用本方法和《SF/Z JD0107005-2016血液、尿液中238种毒(药)物的检测 液相色谱-串联质谱法》对样品进行对比检测,结果见表3。本方法和应用司法技术规范检测的相对偏差在10%之内,可作为司法技术规范的补充检测方法或者确证方法。

表3 本方法和司法技术规范方法的结果对比
本研究应用高分辨质谱技术对干血斑样本中22种镇定安眠药成分进行了测定,干血斑样品经过乙腈提取后,应用基质分散固相萃取法进行净化。结果表明:22种被测物质在线性范围、检出限、定量限、回收率以及稳定性等方面符合方法学要求,该方法样本存储方便,保存时间长,前处理操作简单,为临床药物研究和法医毒物分析提供了新的技术支持。本研究仅选择了两种基质分散固相萃取剂,没有开展对于不同指标类型血,以及不同干血斑产品的适用性方面优选工作,今后研究中,可以从这些方面进行更多的尝试。干血斑-质谱法在精准医学检测、药物研究、疾病诊断等方面具有良好的应用前景。
猜你喜欢 噻嗪水溶液内标 气相色谱内标法测洗涤剂中的甲醇口腔护理用品工业(2021年4期)2021-11-02氢氯噻嗪联合替米沙坦用于高血压治疗的有效性分析世界最新医学信息文摘(2021年12期)2021-06-09厄贝沙坦氢氯噻嗪片在高血压临床治疗中的应用及不良反应状况中华养生保健(2020年10期)2021-01-18厄贝沙坦氢氯噻嗪、叶酸对H型高血压伴舒张性心衰疗效观察研究中华养生保健(2020年5期)2020-11-16水溶液中的离子平衡图像易错题分析中学生数理化(高中版.高二数学)(2020年2期)2020-04-21判断电解质水溶液酸碱性的简单模型中学化学(2019年2期)2019-07-08GC内标法同时测定青刺果油中4种脂肪酸中成药(2018年6期)2018-07-11水溶液中离子平衡的核心考点及复习策略广东教育·高中(2017年9期)2017-09-27DMAC水溶液乙酸吸附分离过程浙江大学学报(工学版)(2016年9期)2016-06-05核磁共振磷谱内标法测定磷脂酰胆碱的含量中国粮油学报(2016年5期)2016-01-23推荐访问:基质 高效 色谱