UGT2B28、FABP5、CYP2C9基因在脂肪酸代谢异常所致大鼠肝纤维化发生中的作用
来源:优秀文章 发布时间:2022-12-04 点击:
宋敬茹 武超 曹红燕 谢咚 王铮 刘璐 王丹 孙明瑜 边艳琴
二甲基亚硝胺(Dimethylnitrosamine,DMN)诱发的肝纤维化大鼠模型已成为研究肝纤维化及肝硬化的经典动物模型[1,2]。研究证实,肝星状细胞活化后转化为肌成纤维细胞分泌大量细胞外基质,导致细胞外胶原沉积增多[3]。前期研究采用全基因芯片结合生物信息学分析技术,发现脂肪酸代谢异常是DMN诱导肝纤维化发生的另一重要原因[2]。本研究在前期研究基础上,对生物信息分析结果深入挖掘,以期阐明脂肪酸代谢异常引起大鼠肝纤维化发生的重要机制。
一、实验动物与材料
(一)动物 Wistar大鼠,雄性,体质量(200±15)g,清洁级,购自中国科学院上海实验动物中心。动物合格证:SCXK(沪)2008-0003。于上海中医药大学实验动物中心进行饲养、造模及观察,自由饮食。
(二)试剂及仪器 甲醛、甲醇、中性树胶、二甲苯、无水乙醇等均购自国药集团化学试剂有限公司;
DMN购自和光纯药工业株式会社;
石蜡购自德国Leica公司;
天狼猩红购自美国Sigma公司;
基因检测及分析所需试剂及仪器设备均由上海生物芯片公司提供。
二、实验方法
(一)造模方法 参照Ala-Kokko方法造模[4]。模型组大鼠以10 mg/kg剂量于每周前3天连续腹腔注射0.5% 的DMN溶液(以0.9% NaCl稀释),持续共4周,对照组大鼠腹腔注射等量的0.9% NaCl。分别在造模2周末、4周末杀鼠取材评估组织病理变化。
(二)样品的采集与处理 在实验2周末和4周末,采用3%戊巴比妥钠,以2 ml/kg剂量腹腔注射麻醉后,打开腹腔,经下腔静脉采血。待血液抽净后,摘取肝脏,然后选取肝脏最厚一叶作为目标组织,切取1.0 cm×1.0 cm肝组织2块,4%甲醛固定,剩余肝组织EP管中-80℃保存备用。组织标本经过脱水、包埋、切片处理后,进行HE和天狼猩红染色[5],观察组织病理学变化。采集血液4℃静置3 h后, 以3000 r/min离心15 min,分离血清,-80℃保存备用。ALT、AST、TC和FFA检测按照试剂盒说明书进行。
(三)肝组织基因芯片杂交实验 采用Trizol试剂盒提取总RNA→纯化→合成cDNA→纯化,参照BioArray(TM)HighYield(TM)RNA转录标记试剂盒方法合成cRNA→将cRNA片段杂交于Afymetrix Rat2302.0芯片→洗脱→染色→扫描芯片。基因芯片:Afymetrix Rat2302.0基因表达谱芯片,共载基因探针31099个。
(四)基因芯片的数据处理、差异基因筛选和生物信息分析 基因芯片的数据处理、差异基因筛选和生物信息分析参照文献[6]。利用R语言进行通路富集分析,利用 Cytoscape 3. 8. 0绘制差异基因参与的脂代谢信号网络图。
三、统计学分析

一、各组大鼠肝组织病理变化
HE染色结果提示,与对照组比较,造模2周时,肝组织内可见大量炎细胞弥漫分布于肝窦内,肝脏结构开始出现紊乱;
随着造模时间延长,肝组织开始出现广泛的肝细胞坏死、肝小叶结构消失。胶原沉积在中央静脉和门脉区并开始向肝窦蔓延是肝纤维化形成的标志。天狼猩红染色结果示,相较于对照组,造模2周组位于中央静脉及门脉区域的结缔组织明显增多,并向肝窦持续蔓延,形成不完全间隔,提示肝纤维化形成;
造模4周时胶原纤维明显较2周时增多、增厚、变粗,相邻的纤维间隔之间彼此连接、并包绕肝小叶形成假小叶,肝脏结构被改变,胶原沉积明显增多,提示肝纤维化已向肝硬化转变,造模成功。见图1。

A、B、C:HE染色×200;
D、E、F:天狼猩红染色×100
二、各组大鼠肝功能与血脂变化
随着造模时间的延长,大鼠血清ALT、AST呈现递增性上升,TG先升高后降低,FFA含量先降低后升高。如表1所示,与对照组比较,4WDMN组大鼠血清ALT、AST酶活性明显升高,且差异有统计学意义(P<0.05);
2WDMN组大鼠血清TG升高,但差异无统计学意义(P>0.05)(考虑是样本离散度太大);
2WDMN组大鼠血清FFA明显降低,且差异有统计学意义(P<0.05);
4WDMN组大鼠血清TG与FFA含量与对照组比较差异无统计学意义(P>0.05)。
三、差异基因分析
选取差异表达大于2倍的基因,并比对其中共同存在的差异基因。2WDMN组与对照组比较显著差异基因数目共有133个,4WDMN组与对照组比较显著差异基因共有98个,两组共同的差异基因共有55个。55个共同差异基因中,上调的基因有40个,下调的基因有15个。在上调基因中,UGT2B28、FABP5为变化最为显著基因,在2WDMN中变化差异倍数均大于5倍,UGT2B28同时在4WDMN组中变化差异倍数也大于5倍;
在下调的基因中,CYP2C9、STAC3、MLC1为变化最为显著基因,在2WDMN、4WDMN中变化差异倍数均大于4倍。
四、共同差异基因参与生物学功能及信号通路
分析55个共同差异基因参与调控的生物学功能及信号通路,对共享基因功能富集,根据Pvalue值从高到低排列,结果显示共享基因参与的前10条通路,其功能主要涉及蛋白酶结合、细胞外基质合成、脂肪酸代谢等方面,见图2。其中,参与脂肪酸代谢功能的通路共有5条, CYP2C9、FABP5、UGT2B28参与了脂代谢所有信号通路,见图3,提示这些基因在DMN诱导脂代谢形成中发挥重要作用。
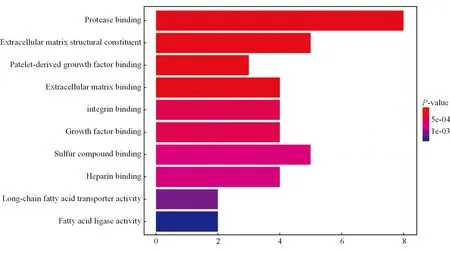
图2 共享差异基因功能富集分析

图3 差异基因参与的脂代谢信号网络图
脂质代谢紊乱与肝星状细胞激活相关,可促进肝纤维的发生发展[7,8]。本研究采用全基因芯片技术,发现差异基因主要参与脂代谢、炎症、免疫和血管新生的调控,可能参与了肝纤维化的发生发展。DNA 的损伤修复贯穿于肝纤维化的发生、发展过程,脂肪酸的代谢紊乱在肝纤维化向肝硬化动态演变的过程中发挥了重要作用。以脂肪酸合成增加为主的脂代谢紊乱,早于肝脏炎症反应,是肝纤维形成的机制之一[9]。

表1 各组大鼠血清肝功能及血脂变化(±s)
全基因芯片深入挖掘后,3个基因调控明显。在下调基因中,随着肝纤维化的加重,CYP2C9逐渐下调。CYP2C9是细胞色素P450家族2亚家族的C型成员之一,与药物代谢以及类固醇、脂肪酸的合成相关[10,11]。CYP2C9的表达水平可以反映肝脏的功能,其在慢性肝病患者中较正常人下调[12]。在肝硬化进展之前,CYPS活性被选择性地改变,肝脏疾病会不同程度地影响CYP亚型的活性[13]。CYP2C9蛋白在肝硬化、肝癌患者中表达均降低,CYP2C9高表达的肝癌患者生存率较高[14]。CYP2C9的下调提示肝纤维化发生发展过程中,肝功能的受损程度加重。研究提示在DMN诱导的肝纤维化大鼠模型中,花生四烯酸代谢紊乱可能促进肝纤维化的发生发展[5]。
在上调基因中,共享差异基因FABP5在2周、4周模型组中明显上调大于5倍。FABP5是细胞内脂肪酸结合蛋白家族之一,与硬脂酸和亚油酸可以高度亲和,参与FAA的摄取、结合、转运和代谢 ,参与炎症反应和能量代谢[15]。同时与肿瘤的发生相关,在HCC、肝内胆管细胞癌等显著上调[16,17]。研究发现,FABP5在LPS诱导的急性炎症反应中起调节作用[18]。FABP5在巨噬细胞中高表达,FABP5的沉默对LPS诱导的肝损伤具有保护作用,并有利于抗炎反应,FABP5-PPARγ信号通路可能促进HSC激活[19]。肝纤维的发生发展过程中,脂肪酸代谢紊乱发生在炎症之前,并可能加重炎症的发展。FAPB5在肝纤维发生的前期主要参与脂肪代谢紊乱,后期主要参与炎症反应。
在上调基因中,共享差异基因UGT2B28在2周、4周上调均大于5倍。UGT2B28是UGT2B亚家族一员,负责胆汁酸和性激素的代谢[20]。UGT2B28基因变异与肝癌的发病年龄、复发、远处转移和死亡有关[21]。本研究发现UGT2B28在肝纤维化形成和发展的过程中上调,提示UGT2B28可能是肝纤维化加剧的预测因子之一。
综上,肝纤维发生的早期存在明显的脂肪代谢紊乱,以基因转录水平调整的脂肪酸紊乱,可能是逆转肝纤维化向肝硬化转化的策略,可以为进一步评估肝纤维化的疗效奠定基础。
猜你喜欢 造模纤维化通路 DJ-1调控Nrf2信号通路在支气管哮喘中的研究进展中国现代医生(2022年19期)2022-11-04变应性鼻炎中促炎信号通路与非促炎信号通路的研究进展*按摩与康复医学(2022年19期)2022-09-27线粒体自噬在纤维化疾病中作用的研究进展中华实用诊断与治疗杂志(2022年1期)2022-08-31AngⅡ激活P38MAPK信号通路在大鼠NSAID相关小肠损伤中的机制研究中国现代医生(2022年19期)2022-08-25炎性及心肌纤维化相关标志物在心力衰竭中的研究进展中国现代医生(2022年21期)2022-08-22恩替卡韦联合安络化纤丸治疗慢性乙型肝炎肝纤维化的研究中国药学药品知识仓库(2022年9期)2022-05-23肝纤维化防治面临的挑战家庭医学·下半月(2022年3期)2022-04-07蛋鸡输卵管炎造模方法的筛选与验证家禽科学(2021年10期)2021-11-22SD大鼠哮喘模型建立方法及评价的比较研究世界中医药(2019年11期)2019-09-10一过性食管下括约肌松弛动物模型的建立及其机制探讨中国现代医生(2019年4期)2019-04-10推荐访问:脂肪酸 代谢 所致