窄治疗指数药物利伐沙班片在中国健康受试者中的生物等效性研究
来源:优秀文章 发布时间:2022-12-02 点击:
王铸辉,阳晓燕,张行飞,阳国平,黄洁,邹婵(.湖南省药品审评与不良反应监测中心,长沙 4003;
.中南大学湘雅三医院临床试验研究中心,长沙 4003)
利伐沙班直接通过抑制凝血因子Ⅹa可中断凝血瀑布的内源性和外源性途径,抑制凝血酶的产生和血栓形成。目前,利伐沙班是血栓栓塞性疾病推荐的一线用药[1-5]。与华法林需频繁监测患者国际标准化比值(INR)相比,利伐沙班的治疗依从性更好。利伐沙班于2008年在加拿大和欧盟获得上市批准,2009年在澳大利亚及中国获批,2011年在美国获批。本研究旨在评价南京恒生制药有限公司生产的利伐沙班片与参比制剂在中国健康受试者的生物等效性及安全性,为国内利伐沙班仿制药上市审批提供依据。
利伐沙班片受试制剂(T,规格:20 mg/片,批号:191201,含量:101.2%,南京恒生制药有限公司);
利伐沙班片参比制剂(R,商品名:拜瑞妥,规格:20 mg/片,批号:BJ45597,含量:101.1%,Bayer AG);
利伐沙班(对照品,纯度:99.7%,批号:ITM10082849,上海陶素生化科技有限公司);
Rivaroxaban-d4(内标,纯度:98%,批号:9-JHY-173-5,Toronto Research Chemicals);
甲醇、乙腈、甲酸(Merck 公司);
二甲亚砜(DMSO,国药集团);
水为超纯水。HPLC30-AD超快速液相系统(日本Shimadzu);
AB Sciex API 5500质谱仪(美国应用生物系统公司)。
2.1 受试者选择
入选标准:年龄18~60周岁,男女均可;
体质量指数(BMI)19.0~26.0 kg·m-2,男性受试者体质量≥ 50.0 kg,女性受试者体质量≥45.0 kg。试验前签署知情同意书,并对试验内容、过程及可能出现的不良反应有充分了解;
志愿者能够和研究者进行良好的沟通,并且理解和遵守本项研究的各项要求。
排除标准:患有需排除的疾病,包括但不限于心血管系统、内分泌系统、神经系统、血液系统、免疫系统(包括个人或家族史遗传性免疫缺陷)、精神病、代谢异常、恶性肿瘤、淋巴组织增生性疾病等病史且研究者认为目前仍有临床意义者;
有食物、药物过敏史,尤其对利伐沙班片及辅料中任何成分过敏者;
有罕见的遗传性半乳糖不耐受、Lapp乳糖酶缺乏症或葡萄糖-半乳糖吸收不良(如喝牛奶腹泻)者;
既往患有任何增加出血性风险或凝血功能障碍疾病者;
在服用研究药物前3 d进食可能影响药物体内代谢的食物者(包括葡萄柚或葡萄柚产品、火龙果、芒果、柚子、橘子等);
试验前1个月内使用过任何抑制或诱导肝脏药物代谢的药物者;
生命体征异常者(收缩压<90 mmHg或>140 mmHg,舒张压<50 mmHg或>90 mmHg;
心率<50 次·min-1或>100 次·min-1)或体格检查、心电图、实验室检查异常有临床意义者(以临床医师判断为准);
研究者认为依从性差或者存在不适合参加试验因素的受试者。
本试验经中南大学湘雅三医院伦理委员会批准(批件号:第20025号),所有受试者均签署知情同意书。空腹组筛选118例,共有27例健康受试者完成试验,包括2例女性和25例男性,平均年龄为(25.8±5.21)岁,平均体质量为(63.31±7.13)kg,平均BMI为(22.17±1.93)kg·m-2;
餐后试验筛选99例,共有28例健康受试者完成试验,包括5例女性和23例男性,平均年龄为(24.4±5.37)岁,平均体质量为(62.29±7.30)kg,平均BMI为(22.61±2.17)kg·m-2。
2.2 试验设计
按照FDA的利伐沙班片生物等效性指南,本试验在健康成年志愿者中进行单中心、单剂量、随机、开放、两制剂、四周期全重复交叉的生物等效性试验。其中空腹、餐后受试者均为 28 例,受试者按筛选号顺序随机分为两个给药序列组(T-R-T-R 组 /R-T-R-T 组),在四周期分别口服20 mg受试制剂T与参比制剂R,清洗期为7 d。给药方法:空腹或者高脂餐后30 min,240 mL温水送服。服药前1 h及服药后1 h内禁止饮水,服药前禁食至少10 h,服药后4 h内禁食,服药后4、10 h统一进食标准午餐和晚餐。分别在给药0时(于给药前60 min内)和给药后0.25、0.5、0.75、1、1.5、2、2.5、3、3.5、4、5、6、8、12、24、36、48 h采血,每次采集约4 mL。血样采集至EDTA-K2抗凝剂中,混匀后即刻置于冰浴中暂存,1 h内将血样在约4℃低温条件下离心(1700 g)约10 min,取分离后的血浆至检测管中和备份管中,离心后的血浆样品在采血后2 h内置于-20℃低温冰箱,并于每周期采血结束后转移至超低温冰箱保存,直至样品转运。
2.3 血浆样本处理与测定
2.3.1 色谱条件 色谱柱:ACE 3 C18-AR(50 mm×2.1 mm,5 μm);
流动相 A:0.4%甲酸水溶液(pH 3.2),流动相B:0.1%甲酸乙腈溶液,梯度洗脱(0.00~1.20 min,35%B;
1.20~1.30 min,35%~100%B;
1.30~1.80 min,100%B;
1.80~1.90 min,100%~35%B;
1.90~3.00 min,35%B);
流 速:0.6 mL·min-1;
柱 温:40℃;
自动进样器温度:4℃;
进样量:5.00 μL。
2.3.2 质谱条件 正离子电喷雾离子化(ESI);
电离模式;
多反应监测模式(MRM)扫描模式;
离子源喷雾电压为 5.0 kV,离子源温度为500℃,入口电压10 V,碰撞气9 unit,出口电压13 V,离子源GS1为50 psi,离子源GS2为55 psi。用于定量分析的离子反应为利伐沙班m/z436.2 →145.0,内标Rivaroxaban-d4m/z440.3→145.0。
2.3.3 血浆样品处理 精密移取样本100 μL(空白样品和内标空白样品取100 μL空白人血浆)至96 孔板中;
加入20 μL内标工作液,混匀;
加入600 μL乙腈,加盖96孔板盖涡旋3 min混匀,3200 g、4℃离心5 min;
取400 μL上清液至另一96孔板中,40℃氮气流吹干;
加入200 μL复溶液[0.4%甲酸水溶液(pH3.2)∶0.1%甲酸乙腈(65∶35)],加盖96孔板盖涡旋3 min,即得。
2.4 方法学考察与评价
2.4.1 特异性 取6例受试者的空白血浆,按照“2.3.3”项下方法处理后进样,血浆内源性物质的峰面积均小于利伐沙班最低定量下限(LLOQ)(1 ng·mL-1)峰面积的20%,小于内标峰面积的5%。结果表明,血浆内源性物质不干扰待测物和内标的测定,该方法的特异性良好(见图1)。
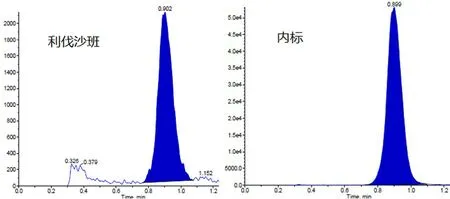
图1 血浆中分析物和内标的典型色谱图Fig 1 Typical chromatogram of analyte and internal standard in plasma
2.4.2 标准曲线与LLOQ 配制标准曲线血浆样品(1、2、10、50、100、250、400、500 ng·mL-1),按“2.3.3”项下方法操作后进样分析,建立利伐沙班的标准曲线。以质控样品质量浓度为横坐标(X,ng·mL-1),利伐沙班与内标的峰面积比值为纵坐标(Y),采用加权回归法进行回归,权重因子为 1/χ2。结果表明利伐沙班在1~500 ng·mL-1内与峰面积线性关系良好,其回归方程为Y=0.402X+0.0021(R2=0.9986),LLOQ为1 ng·mL-1。
2.4.3 精密度与回收率 配制利伐沙班LLOQ质量浓度(1 ng·mL-1)、低质量浓度(LQC,3 ng·mL-1)、中质量浓度1(MQC1,20 ng·mL-1)、中质量浓度2(MQC2,200 ng·mL-1)、高质量浓度(HQC,375 ng·mL-1)的质控样品,每个浓度6份,重复3次,按“2.3.3”项下方法处理后进样分析,结果批内和批间精密度(RSD)均<5%,准确度在99.4%~101.8%。用LQC、MQC2、HQC 3个质量浓度样品,比较经提取与未经提取利伐沙班和内标的各自峰面积比值,结果低、中、高质量浓度的平均提取回收率分别为96.34%、93.45%、92.01%,RSD分别为1.8%、0.30%、0.56%。
2.4.4 基质效应 取6例受试者的空白血浆,分别考察LQC、MQC2、HQC 3个质量浓度的利伐沙班在基质离子存在时色谱峰响应和在基质离子不存在时色谱峰响应的比值。内标归一化基质因子为待测物的基质效应除以内标的基质效应。结果3个质量浓度的利伐沙班内标归一化基质因子均值分别为100%、100%和101%。
2.4.5 稳定性 利伐沙班全血样品在冰浴条件下放置1 h后离心,稳定性良好;
利伐沙班血浆样品在室温和冷藏下放置24 h、在-30~-10℃至室温以及-90~-60℃至室温3次冻融循环、在-30~-10℃以及-90~-60℃条件下长期冰冻40 d稳定性良好;
利伐沙班血浆样品处理过程中在1~9℃条件下放置 24 h稳定性良好、处理后样品1~9℃条件下放置123 h稳定性良好。
2.5 药动学参数计算及生物等效性判定
药动学参数采用非房室模型,使用Phoenix WinNonlin8.0(美国Pharsight公司)软件和SAS 9.4进行统计分析。主要药动学参数AUC0~t、AUC0~∞、Cmax经自然对数转换后进行多因素方差分析(ANOVA)、双单侧t检验和90%CI分析。当受试制剂T和参比制剂R药动学参数Cmax、AUC0~t、AUC0~∞经对数转换后几何均值比的90%CI均落在80.00%~125.00%,且受试制剂T和参比制剂R个体内标准差比值的90%CI上限≤2.5时[6],则认为两制剂具有生物等效性。
2.6 血药浓度-时间曲线
空腹组完成随机化28例,1例受试者于第一周期给药前退出,共有27例受试者完成试验。餐后组完成随机化28例,1例受试者于第四周期给药前退出(已完成3周期给药盒采血),共有27例受试者全部按计划完成试验。对完成血样采集的样本进行血药浓度检测,空腹组和餐后组平均血药浓度-时间曲线见图2和图3。

图2 空腹服用受试制剂和参比制剂的平均药时曲线图Fig 2 Mean plasma concentration-time curve of 27 subjects after an oral dose of rivaroxaban tablets under fasting condition

图3 餐后服用受试制剂和参比制剂的平均药时曲线图Fig 3 Mean plasma concentration-time curve of 28 subjects after an oral dose of rivaroxaban tablets under fed condition
2.7 药动学参数
空腹组27例受试者和餐后组28例受试者均纳入药动学参数集(PKPS)分析,结果见表1。
表1 空腹组和餐后组受试者单次口服利伐沙班片受试制剂和参比制剂的药动学参数(±s)Tab 1 Main pharmacokmetic parameters of rivaroxaban tablets of the test or the reference preparation in both fasting and fed conditions (±s)

表1 空腹组和餐后组受试者单次口服利伐沙班片受试制剂和参比制剂的药动学参数(±s)Tab 1 Main pharmacokmetic parameters of rivaroxaban tablets of the test or the reference preparation in both fasting and fed conditions (±s)
注:*表示tmax采用中位数(最小值,最大值)的形式描述。Note:*means tmax is described in terms of median(minimum,maximum).
组别 制剂 tmax/h* Cmax/(ng·mL-1) AUC0~t /(h·ng·mL-1)AUC0~∞/(h·ng·mL-1) t1/2/h空腹组(n=27) 受试制剂T 1.50(0.50,4.00) 195.28±46.01 1490.58±356.92 1609.00±394.74 10.12±5.88参比制剂R 2.00(0.50,4.00) 197.48±52.63 1439.72±372.20 1538.39±386.49 10.37±6.82餐后组(n=28) 受试制剂T 2.50(1.00,4.50) 402.05±85.67 2776.66±756.91 2801.41±755.55 4.52±0.95参比制剂R 2.50(0.50,4.50) 394.42±65.29 2728.24±801.16 2760.30±805.05 4.86±1.36
2.8 生物等效性评价
受试制剂T和参比制剂R的Cmax、AUC0~t、AUC0~∞对数转换后的几何均数比值GMR的90%CI均落在 80.00%~125.00%,双单侧t检验结果与置信区间法结果一致,受试制剂T和参比制剂R个体内标准差比值的90%CI上限≤2.5,说明两制剂在本实验空腹和餐后给药条件下具有生物等效性[6],详见表 2、3。
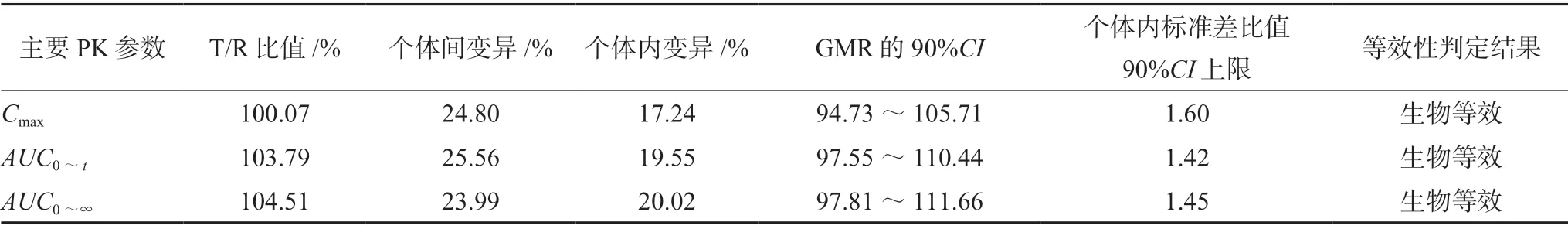
表2 空腹口服受试制剂与参比制剂的主要PK参数生物等效性评价结果Tab 2 Main pharmacokmetic parameters of rivaroxaban tablets of the test or the reference preparation in fasting condition

表3 餐后口服受试制剂与参比制剂的主要PK参数生物等效性评价结果Tab 3 Main pharmacokmetic parameters of rivaroxaban tablets of the test or the reference preparation in fed condition
2.9 安全性评价
空腹组27例受试者全部按计划完成给药、采血和安全性评价,并进入安全性分析集。共有12例受试者发生17例次不良事件。服用受试制剂T期间有5例(18.5%)受试者发生了7例次不良事件,服用参比制剂R 期间有8例(29.6%)受试者共发生了10例次不良事件。不良事件为上呼吸道感染、尿白细胞酯酶阳性、潜血阳性、血三酰甘油升高、血尿酸升高,均为轻度不良事件,未进行任何治疗,均恢复正常。
餐后试验 27 例受试者全部按计划完成给药和采血,1例受试者完成了3个周期给药和采血,共有28例受试者进入安全分析集。结果15例受试者发生22例次不良事件,其中服用受试制剂T期间有8例(28.6%)受试者发生了11例次不良事件;
服用参比制剂R期间有10例(35.7%)受试者发生了11例次不良事件。不良事件为上呼吸道感染、咽炎、尿白细胞阳性、尿蛋白检出、心率升高、血三酰甘油升高、腹泻。有1例受试者发生中度不良事件“上呼吸道感染”,采取治疗措施后恢复正常。其余均为轻度不良事件,未进行任何治疗,均恢复正常。
本试验研究期间两药安全性相近,未发生严重不良事件,故受试者在空腹及餐后给药条件下口服受试制剂T和参比制剂R 20 mg后临床安全性良好。
利伐沙班能选择性地阻断Ⅹa因子的活性位点起到抗凝作用,其血药浓度的微小变化可能导致血栓或出血,进而危及生命,为窄治疗指数药物[1-5]。与一般化学药物相比,窄治疗指数药物进行生物等效性评价时,应采用更严格的等效性判定标准,以保证有效性和安全性。FDA在利伐沙班片生物等效性指南中建议,利伐沙班片的生物等效性统计分析应采用平均生物等效性ABE法(80%~125%),并比较受试者服用受试制剂T和参比制剂R的个体内变异,且两药个体内变异标准差比值的90%CI上限应≤2.5[6-10]。
因此,本次研究采用四周期全重复交叉的设计,以获得两药的个体内变异。个体内变异系数取24%,预计受试制剂与参比制剂药代参数均值差异为10%,α=0.05,β=0.2(把握度1-β=80%),利用软件PASS 11.0进行样本量计算,计算结果表明纳入不少于26例能够可靠的进行等效性判断,考虑脱落,将样本量定为28例。结果显示,本次研究利伐沙班个体内变异在14.11%~20.02%,Cmax、AUC0~t和AUC0~∞个体内变异标准差比值的90%CI。
本次研究中,与其他文献中利伐沙班等效性研究结果相似,利伐沙班在空腹组和餐后组中的部分药动学参数(tmax、Cmax、AUC、t1/2)差异较大。餐后组中的tmax、Cmax、AUC较大,可能原因是高脂餐后给药,会延长药物的释放,增大药物在体内的吸收,从而增大药物在体内的暴露。在临床治疗上,该药动学性质可以为剂量调整提供依据,t1/2差异较大,可能原因是餐后组PK曲线消除相采样点数量较少,药时曲线不平滑,导致计算出来的t1/2与空腹组相比差异较大,是本次研究的不足之处。如果在餐后组增加18 h和30 h采样点,就能得到更平滑的消除相曲线。
猜你喜欢 药动学血浆受试者 浅谈新型冠状病毒疫情下药物Ⅰ期临床试验受试者的护理世界最新医学信息文摘(2022年43期)2022-11-19抗菌药物的药动学和药效学参数对临床用药的意义中国药学药品知识仓库(2022年5期)2022-04-11你真的了解献血浆是怎么回事吗?康颐(2020年15期)2020-11-10青娥丸方有效成分药动学药效学相关性研究中国中药杂志(2016年23期)2017-04-07尼莫地平/川芎嗪双载药纳米粒的体内药动学和脑组织分布研究中国中药杂志(2016年22期)2017-02-13适度惩罚让人进步现代家长(2016年11期)2016-12-05重要的事为什么要说三遍?党的生活(黑龙江)(2016年8期)2016-08-15醋酸艾司利卡西平片在比格犬体内的相对生物利用度上海医药(2016年1期)2016-02-22逼中小学生卖血的背后中国新闻周刊(2014年32期)2014-05-14简易姿势测定法祝您健康(1983年1期)1983-12-29推荐访问:中国 药物 受试