深度学习下主流染色体分类算法的性能评估
来源:优秀文章 发布时间:2023-04-16 点击:
易序晟,尹爱华,黄杰晟,彭璟,陈汉彪,郭莉,林成创,李双印,赵淦森*
1. 华南师范大学计算机学院,广州 510631;
2. 广州市云计算安全与测评技术重点实验室,广州 510631;
3. 华南师范大学唯链区块链技术与应用联合实验室,广州 510631;
4. 广东省妇幼保健院,广州 511400
根据2010年世界卫生组织发布的《出生缺陷报告》,先天性疾病是常见疾病,全世界因先天性异常而死亡的人数约为260 000人,占所有新生儿死亡病例大约7%。中国的出生缺陷儿绝对数量巨大。据《中国出生缺陷防治报告(2012)》(中华人民共和国卫生部,2012)报告显示,我国出生缺陷发生率在5.6%左右。先天性疾病和出生缺陷可以造成自然流产和死产,是导致新生儿死亡和残疾的一个重要原因。它们可以危及生命,导致长期残疾,从而对个人、家庭和社会造成不利影响。因此,针对出生缺陷诊断的科学研究具有重要价值(Abid和Hamami,2018)。
染色体包含着人类遗传信息,常用于分析人类遗传病。一般来说,人体细胞中在正常情况下包含46条染色体,其中由22对常染色体(1—22类)和1对性染色体(女性为两条X性染色体,男性为一条X染色体和一条Y染色体)构成(林成创 等,2020)。染色体核型化分析是产前诊断和遗传疾病诊断的重要且常用方法,是临床上遗传疾病诊断的金标准。如图1所示,染色体核型化分析是指从细胞分裂中期的显微镜图像中分割得到染色体实例后,凭借ISCN(international system for human cytogenetic nomenclature)标准(Shaffer等,2013),最终形成染色体核型图的过程。

图1 染色体核型分析示例
医学影像是现代医学最重要的临床诊断和鉴别诊断方法,也是计算机辅助诊断的重要依据(田娟秀 等,2018)。染色体分类作为医学影像处理的子任务,是临床遗传疾病诊断的重要方法,通常由熟练的细胞遗传学专家手工完成。受染色体非刚性特性影响,同一类型的染色体常具有完全不同的形状和方向,给染色体分类研究带来巨大挑战。染色体分类是染色体核型化分析中最复杂也是最容易出错的中间环节,对临床医生出具产前诊断结果有着很好的借鉴价值。因此,染色体分类是一类极具挑战性且值得深入研究的热点问题。
在深度学习兴起之前,染色体分类技术主要分为两大技术路线,基于人工神经网络(artificial neural network,ANN)的技术路线(Kubola和Wayalun,2018;
Popescu等,1999;
Moradi等,2003;
Oskouei和Shanbehzadeh,2010)和基于概率神经网络(probalilistic neural network,PNN)的技术路线(Poletti等,2011;
Rungruangbaiyok和Phukpattaranont,2010;
Khan等,2012)。第1类方法主要采用多层感知机(multilayer perceptron,MLP)提取染色体特征,但有一个复杂的特征筛选过程;
第2类方法的精度较低,所需的训练时间较短。早期染色体分类方法有着复杂的特征选择过程,手工设计的特征提取器有固有的局限性,无法全面涵盖图像的低级像素特征,更加难以设计出高层抽象的特征,导致染色体的错误分类(金旭 等,2020)。
随着以深度学习为代表的人工智能技术的快速发展,采取深度学习技术的医学影像分析成为研究热点,吸引众多学者投入染色体分类的研究。Sharma等人(2017)和Swati等人(2017)分别较早地提出基于改进卷积神经网络和基于改进孪生网络的染色体分类方法,在各自选择的数据集中取得了86.7%和84.6%的分类准确率。Lin等人(2020b)提出残差神经网络变体模型和自适应接口(image adaptive interface,IAI)的染色体分类方法。其中,自适应接口有效解决了染色体图像尺寸输入不一致问题。在作者人工标注的2 990条染色体图像数据集中,测试准确率为95.98%。Zhang等人(2021)提出了结合交错和多任务学习网络的染色体分类和校正模型,按3个阶段进行特征学习。在作者收集的数据集上进行性能评估,准确率为98.1%。
染色体分类任务研究取得了一定进展,但基于深度学习的染色体分类研究存在滞后性,且暴露出一些问题。1)难以收集到大规模带标签的染色体数据。数据量和数据质量都在数据获取过程中存在明显的人员和技术阻碍,存在数据壁垒。标准化数据往往采取人工标注方式,存在着标注难度大、标注信息难以获取以及高成本等问题。2)模型性能的好坏与训练数据量密切相关。深度学习需要大量的训练数据,目前染色体分类研究大量依赖高成本标注背景下的染色体小样本数据。虽然目前染色体分类算法已经取得了一些进展,但大部分是在轻量化数据集上得到的不同水准的分类性能,训练数据不够充分,导致现有模型可能处于低层次精度水平。3)模型难以验证,成果转化难。
存在上述问题的根本原因是数据量缺乏。针对上面的问题,本文尝试从以下3个方面开展研究工作。1)针对第1个问题,构建目前最大规模的临床染色体数据库。其中,分割染色体实例以及开展的数据标注工作全部由专业临床医生团队全程参与并完成。2)针对第2个问题,在保证数据量的前提下,提供给现有模型足够的训练数据,尝试验证现有模型性能能否得到整体的提升。3)针对第3个问题,在构建好的数据集上展开染色体分类算法之间的性能评估实验,探究不同网络结构之间是否存在性能差异,为临床应用提供对网络模型的初筛建议。
为开展性能评估实验,本文工作从广东省妇幼保健院医学遗传中心获得126 453条染色体样本的临床数据集,从近年基于深度学习的染色体分类的相关研究成果中重点挑选出6种主流染色体分类算法,所有参评模型进行染色体分类识别性能测试(https://github.com/CloudDataLab/ChromesomeBen-chmark)。本文主要贡献如下:1)阐述近5年基于深度学习的染色体分类方法。按网络结构的设计思想和时间发展顺序进行介绍,并给出网络间存在的联系,对相关研究人员有较好的借鉴意义。2)精选6个主流染色体分类模型在大规模临床数据集展开性能评估实验。本次评估实验是在染色体分类研究领域中第一次进行较大规模实验评估,对染色体分类研究具有重要价值。3)在评估实验中,带有不同参数量的模型借助准确率、精确率、召回率和F1值4类评价指标来分析模型在大型数据集上的分类潜力,这对于更加准确地认识深度学习在染色体分类中的分类性能具有重要意义。
染色体具有非刚性的内在性质。在显微镜下染色体形状和方向的多元化对于染色体分类过程带来了巨大挑战(Grisan等,2009)。随着深度学习技术的发展,将深度学习技术用于染色体分类成为研究热点,但确保数据规模和设计出高性能分类模型存在巨大挑战。
1.1 问题定义和描述
染色体分类是将单条染色体图像按照ISCN规则确定所有染色体组号和染色体号的过程,如图2所示。关于染色体组号,ISCN规则(McGowan-Jordan等,2016)将染色体分成7组,从A到G依次编号。其中A组包括1、2、3号常染色体,B组包括4、5号常染色体,C组包括6—12号常染色体以及X性染色体,D组包括13、14、15号常染色体,E组包括16、17、18号常染色体,F组包括19、20号常染色体,G组包括21、22号常染色体和Y性染色体。关于染色体号,ISCN规则中对染色体核型分析的描述如下:在核型图中,常染色体按照长度递减的顺序用数字1到22编号(唯一例外是21号染色体比22号短),性染色体用X和Y表示。
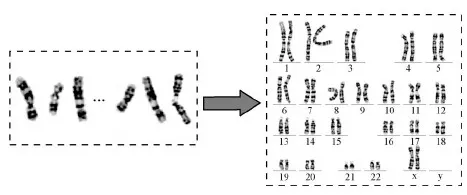
图2 染色体分类过程
染色体分类问题使用形式化公式描述如下:使用X={x1,x2,…,xN}表示包含N个染色体图像样本的数据集,其中xj表示第j个染色体图像样本;
使用Y={y1,y2,…,yN}表示与X数据集对应的染色体真实类别集合, 通常称为ground truth(GT)标签集;
其中yj∈{1,2,…,22,X,Y}表示第j个染色体图像样本的真实类别;
使用F(·)表示一个染色体分类算法或模型,使用y′j=F(xj)表示F(·)算法或模型推理第j个染色体样本的预测类别;
使用Y′={x′1,x′2,…,x′N}表示F(·)的分类结果集合,通常称为Predicted Set测试数据集。
1.2 常用数据库和评价指标
1.2.1 常用数据库
染色体影像依赖显微镜制作。采样过程中,既要保证拍摄的影像完全呈现出细胞中的所有染色体,还需要尽可能提高染色体图像的清晰度,无法同时兼顾成像视野与放大倍数,而且制作染色体图像的过程过于烦琐。现实中收集一个大型且带有标注信息的染色体数据集难度更大,一方面,收集过程费时,还需相关专业领域的知识储备;
另一方面,医疗机构出于隐私保护等医学伦理问题的考虑,基本不公开相应染色体数据。即使收集数据途径得到保证,但将数据集进行开放共享需具备一定的前提条件,即科研团队和医疗机构存在着研究合作关系并需满足患者知情以及严格的数据脱敏等要求。打造高质量的大型公开数据集过程相当具有挑战性。历年染色体分类研究数据集如表1所示。

表1 染色体分类常用数据库
最早公开的数据集是Poletti等人(2008)提出并由细胞学专家手动分割得到的,名为BiolmLab数据集。数据集分为119个文件夹(细胞),每个文件夹包含46条染色体,共5 474条染色体,总大小为10.5 MB(图像格式:BMP,8位/像素)。每幅图像分辨率大小取染色体的最佳分布位置,以保证标注数据的高质量。BiolmLab数据集(http://bioimlab.dei. unipd.it/)对染色体分类有着重要的研究价值。部分数据集如图3所示。Hu等人(2019)从91张核型图片中分割得到4 184条正常染色体图像并制作成公开数据集(https://github.com/Xi-Hu/Chromosome- Images)。其中,图像背景统一扩展为100(宽)×220(高)像素。染色体标记信息体现在文件名中。例如,在“161923.054.K.chr1.1invert_pad.gif”文件名中,“161923.054”是样本编号,K代表核型图像,“chr1.1”代表该图像是成对染色体1号中的一条(另一条染色编号为chr1.2)。Lin等人(2020b)于2020年发布了染色体数据库(https://github.com/ CloudDataLab/CIR-Net), 该数据集包括2 990幅尺寸约为224×224像素的单条染色体图像,每个像素占用的有效比特数为8(8bpp)。

图3 BiolmLab数据集部分情况
手工标注染色体数据是一个费时费力的阶段(周雪雁 等,2004)。因此,收集大型公开可用的染色体数据集过程具有挑战性。值得注意的是,即使数据集极少数开源,但染色体分类的研究从未间断。
1.2.2 评价指标
染色体分类是一个多分类任务,但实际操作时往往将每类染色体都单独作为一个二分类问题,然后再对所有类别进行加权计算。其中任意一组染色体的二分类混淆矩阵如表2所示。对于第j组染色体,TPj表示实际属于类别j的染色体被模型预测为j。FPj表示实际不属于类别j的染色体被模型预测为j。FNj表示实际属于类别j的染色体被模型预测为不是类别j。TNj表示实际不属于类别j的染色体被模型预测为不是类别j。

表2 二分类混淆矩阵
不同的分类算法需要进行客观评估,尤其是量化的评估指标。对于染色体分类问题,更加需要一类具备客观可量化、可解释以及被广泛接受特性的评价指标。常用染色体分类算法的评价指标有准确率(accuracy,ACC)、精确率PPV(positive predictive value)、召回率TPR(true positive rate)和F1分数(F1score)。准确率指每一类染色体在所有染色体中正确分类的比例,可以判断总的分类正确率,常作为评估分类效果的最主要和最常用的指标。精确率也称查准率,用于评估所有被预测为正样本中实际为正样本的概率。精确率考察单个类别的准确率,准确率考察整体的准确率。召回率也称查全率,用于评估实际为正样本中被预测为正样本的概率。F1分数是精确率和召回率的加权调和平均值。分别表示为
(1)
(2)
(3)
(4)
式中,N表示染色体的类别总数。
上述评价指标能够提供分类任务中的评价参考,但在实际使用时需根据任务需求选择不同评价指标进行组合,多方面评价分类算法的准确性和稳定性。
1.3 模型设计
染色体分类网络模型主要分为3种设计思路,即网络内部堆叠卷积层的设计思路、网络内部精细化模块的优化设计思路(如跳跃连接和相似度匹配的思想)以及拼接图像多尺度特征的多任务模型设计思路。针对以上3种设计思路,本文提出了基于深度学习的染色体分类算法的划分框架,如图4所示。具体分为基于卷积神经网络(convolutional neural network,CNN)及其变体的分类模型、基于ResNet和Siamese的分类模型以及自设计的多任务学习分类模型。
图4 深度学习下染色体分类研究
1.3.1 基于CNN及其变体的分类模型
卷积神经网络(CNN)作为深度学习的代表性算法之一,是包含卷积计算和一定深度结构的前馈型神经网络,借助堆叠卷积层层数的网络模型能有效将大数据量的图像降维成小数据量,使原图像信号特征增强,并降低噪音。不同于传统的图像分类方法,基于卷积神经网络的图像分类方法具有很好的表征学习能力,其卷积核参数共享和层次化的核心思想使CNN能对图像像素进行潜在特征学习,较大幅度地提高图像分类效果。
Sharma等人(2017)提出结合众包技术、数据预处理和卷积神经网络(CNN)算法进行染色体分类。该研究将染色体灰度图像集的分割任务众包给专业技术人员,构建了染色体数据集。在此数据集上,按照拉直弯曲染色体、染色体图像归一化和使用Deep CNN模型3个流程将染色体分为24类。Deep CNN模型架构如图5所示,该模型串行了4个卷积模块,对120×120像素的染色体图像多重卷积下采样,自学习训练样本的内部特征。池化层也称下采样层,会压缩输入的特征图,一方面减少了特征数,能显著减少训练参数,另一方面保持了特征的某种不变性(旋转、平移和伸缩等)。模型的训练与测试在1 800条染色体下进行,经过数据预处理操作后,实验表明,Deep CNN模型有较好的性能,准确率为86.7%。

图5 Deep CNN架构的染色体分类模型(Sharma等,2017)
Remya等人(2020)在Kiruthika等人(2018)方法启发下,提出了CNN变体双模型ChromNet1和ChromNet2,如图6所示。ChromNet1是一个包含卷积层和池化层(或称采样层)构成的浅层网络模型。模型选取的3×3卷积核(小的感受野)不仅能使模型专注图像的细节信息,并且可以减缓后续参数量的剧增。激活函数选取线性整流单元(ReLU)而不是Sigmoid函数,能有效应对训练过程中出现的梯度饱和以及梯度消失问题。ChromNet2网络在ChromNet1网络的基础上,增添了多层卷积模块和dropout方法(Srivastava等,2014)。在私有标注的21 423条染色体构成的数据集上进行预训练和性能测试,ChromNet2的分类准确率为91.3%,ChromNet1的分类效果在文献中没有提及。
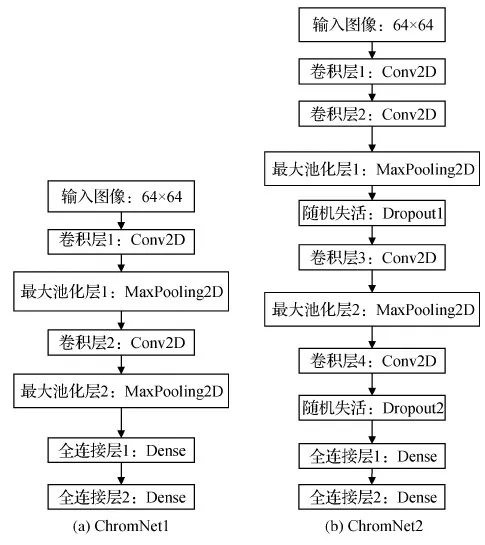
图6 ChromNet1和ChromNet2(Remya等,2020)
减少训练开销的常用策略是权值共享(weight share),这个策略在卷积神经网络中发挥了重要作用。经典Le-Net网络(Lecun等,1998)一经提出就受到了学者们的广泛关注和深入研究,Zhang等人(2018)提出Vanilla CNN网络就借鉴了这一结构。如图7,120×40像素的染色体图像送入Vanilla CNN网络,模型拼接多个卷积层和池化层对输入信号进行加工,然后在连接层实现与输出结果之间的映射。每个卷积层都包含多个特征映射,其中每个特征映射是由多个神经元构成的多维平面,并通过卷积核提取特征。池化层(pooling)的别称为“汇合”层,其目的是基于局部相关性原理进行亚采样,从而既能减少数据量也能留住有效信息。Vanilla CNN网络将原始染色体图像映射成高维特征向量,最后通过一个由120个神经元构成的连接层和输出层连接完成分类。其中,随机失活半数神经元(随机失活dropout=0.5)可以减少神经元之间复杂的共适应关系,增强网络的泛化能力。该方法在包含10 304个染色体图像的私有G带染色体数据集上的分类准确率为92.5%。然而,数据集中样本分布不均衡,仅包含了垂直染色体,所以该模型的分类结果在遇到新的数据集时容易出现染色体的错误分类。
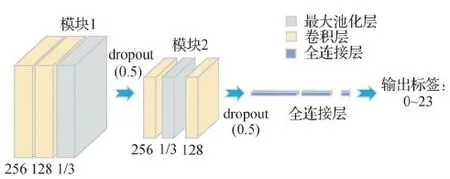
图7 基于Le-Net网络变体的染色体分类模型(Zhang等,2018)

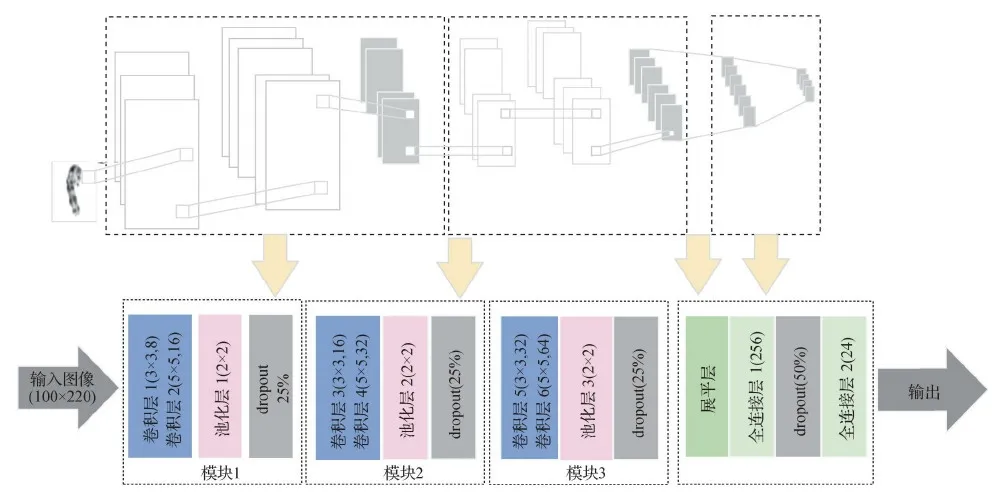
图8 基于VGGNet架构的染色体分类模型(Hu等,2019)
对于如何充分获取图像的多重特征,目前存在着从模型外部拼接不同尺度图像组合后作为输入端的改进思路。例如,将不同形状图像产生的复合信号输入模型以展开训练。李康等人(2020)认为,染色体形状多变性的客观因素和普通CNN模型存在特征提取不充分问题,会带来染色体分类精度不佳的困扰,提出了融合多图像特征的Multi-input CNN模型,借助Mask R-CNN(He等,2017)实现染色体的自动分割,如图9所示。染色体图像经几何优化操作得到优化图像,随后将原始图像和优化图像并行作为Multi-input CNN模型的输入信号。分类过程可精简为残差网络提取特征、融合多层次特征图、使用多层感知机(MLP)进行分类预测3个步骤。数据集源于医院提供的核型图中分割出的染色体实例,共5 000幅128×128尺寸的染色体图像,每张图片包含1条染色体。为了保证Multi-input CNN模型的效果,先训练3个单独的CNN模块直到收敛,然后拼接3个子网络,固定CNN的参数且只训练顶层的分类器,最后解锁整个模型,并在一个很低的学习率(0.000 5)下进行微调,评估结果准确率为95.6%。

图9 Multi-input CNN网络架构(李康 等,2020)
卷积神经网络是一种通过模拟生物大脑皮层结构而特殊设计的含有多隐层的人工神经网络。卷积层、池化层和激活函数是卷积神经网络的核心部分。卷积神经网络通过局部感受野、权重共享和降采样这3种策略,降低了网络模型的复杂度,因此增加网络深度进而提升网络的分类性能是积极有效的一种常规技术方案。
1.3.2 基于ResNet和Siamese的分类模型
为进一步提升网络的分类效果,一些学者从网络内部着手,提出了允许输入信息可以跨越后续网络层并进行传播(即残差网络)和允许两个输入共享编码层(即孪生网络)的基于卷积操作模型。
神经网络一味地增加深度和宽度会面临梯度消失和梯度爆炸导致的网络退化问题,具体表现为网络性能不再随深度增加而提升。为解决此类问题,He等人(2016)提出残差网络,并在ImageNet(Deng等,2009)比赛中取得佳绩。残差网络允许输入信息可以跨越多个隐含层传播,并始终保持信息畅通状态,极大降低了网络复杂度,加速了网络训练过程,同时突破了由网络退化引起的深度限制。
因其通用性与实用性并存,张成成等人(2020)提出了如图10所示的染色体分类模型。该模型由4个残差子模块组成,每个子模块包含两层1×1卷积层(Lin等,2013)和3×3卷积层。将1 225例受检人员的分裂中期染色体图像纳入数据集,并分为训练集735例、内部验证集245例和外部验证集245例;
另取50例染色体图像资料开展人机对抗实验。分类结果显示,模型分类准确率为91.22%。同时,该模型每幅图像测试平均耗时仅需0.06 s,远低于专业医师62.5 s的水准。该方法凭借跳跃连接的思想找最优路径,使分类过程耗时短、效率高,但数据量问题仍未得到有效解决。
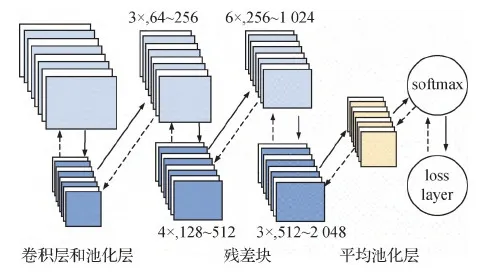
图10 ResNet网络的染色体分类模型(张成成 等,2020)

图11 Res-CRANN网络架构图(Sharma等,2018)
对于如何更进一步提升卷积神经网络性能,目前的主流观点是从网络内部结构着手,通过增加网络深度和宽度来实现,但会出现网络训练参数量大幅增加和训练速度降低问题。为解决上述问题,Szegedy等人(2015)从网中网(Lin等,2013)中得到启发,提出了Inception-V1网络结构。与Lin等人(2013)方法类似,Szegedy等人(2017)使用了类似结构,设计并提出了Inception-ResNet网络模型。
Lin等人(2020b)借鉴了Inception-ResNet模型架构,提出一种多注意网络CirNet进行分类,如图12所示。与传统卷积神经网络不同,该网络以成熟的Inception-ResNet架构作为主干网络,附加深度可分离卷积和卷积核分解等优化策略,并增添了图像增强算法(chromosome data augmentation,CDA)以增强图像特征,同时使用图像自适应接口(image adaptive interface,IAI)将染色体图像规格统一化。CirNet模型多方位了解和推断2 990条染色体,准确率为95.98%,代码和数据集可从GitHub获取。
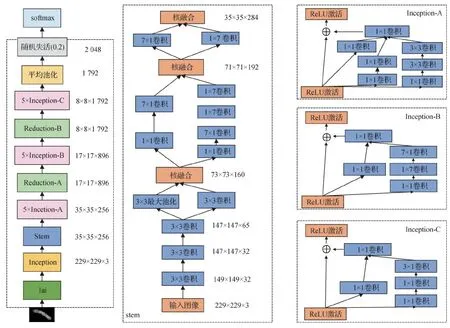
图12 CirNet网络的架构图(Lin等,2020b)
He等人(2016)首次提出,Xie等人(2017)开发的批量残差网络ResNeXt体系结构是一种精简但高度模块化的网络体系结构,它执行一组变换,每个变换基于低维嵌入,其输出通过求和进行聚合。Lin等人(2020a)(MixNet)在ResNeXt的基础上提出一种新颖的MixNet框架,如图13所示。模型可以有效地探测染色体图像的类别信息表征,最终在染色体分类任务中获得了满意的结果。MixNet由双模块构成,染色体编码骨干网络和自适应报头。聚集残差架构构成的染色体编码骨干可以将染色体图像潜在特征通过卷积等方式编码成向量形式,是MixNet的染色体特征编码的核心模块;
自适应报头通过最大池化层捕捉图像关键特征,同时利用全局平均池化层捕捉一般特征。在两个不同数据集下分别训练和测试,测试结果表明,MixNet在包含2 990条染色体的临床G带数据集上的分类准确率为(98.73±0.81)%,在包含5 474条染色体的公共Q带数据集上的分类准确率为(96.50±1.87)%,在两个数据集上准确率均超过95%,且仅需少数迭代(训练时间不到1 h)就能使模型结束训练阶段。

图13 MixNet网络架构图(Lin等,2020a)
上述4种染色体分类研究都是在ResNet网络的基础上改进的模型结构。现有方法常通过各种图像预处理、扩张模型深度和宽度、引入各种优化技术和多模块串行等方法增强算法的鲁棒性和泛化能力,都取得了超过90%的分类效果。孪生网络Siamese最早由Bromley等人(1993)提出,是用来验证支票字迹与银行预留笔迹是否一致的一种方法。在此基础上,涌现出大量针对孪生网络结构的变体模型(Zagoruyko和Komodakis,2015),更有尝试将其应用于计算机视觉领域(Bertinetto等,2016;
Valmadre等,2017)。Swati等人(2017)提出将孪生网络用于染色体图像上的分类算法,使用中轴和众包(straightening via medial axis extraction and crowdsouring,SMAC)以及投影向量(straightening via projection vectors,SPV)的矫正方法来拉直染色体。拉直的染色体输入至由Base-CNN网络组建的孪生网络进行预训练,如图14所示。

图14 孪生网络中使用的Base-CNN架构(Swati等,2017)
孪生网络等同于连体的神经网络,连体通过共享权值实现。在低维空间下,孪生网络可以拉近同类染色体间的空间距离,拉长不同类别染色体之间的空间距离。由于训练数据量缺乏,该模型在染色体分类任务上的准确率为84.6%。
吕丹丹(2019)将孪生网络应用于染色体图像分类中,采用Multi-Siamese架构进行迁移学习以及增添空间金字塔池化层和多级卷积,如图15所示。

图15 Multi-Siamese网络架构(吕丹丹 等,2019)
Multi-Siamese网络模型引入空间金字塔池化层取代网络末端的池化层,解决了对输入数据固定大小的限制。为全面提取染色体特征,模型采用一种多级卷积特征提取结构,该结构能深层次地提取染色体特征,孪生网络模型精度进一步提升。该方法将提取的大小不同的特征图转换成固定大小的特征向量,送入全连接层形成一维向量,最后根据欧氏距离计算特征向量间的相似度。部分参数设定如下:初始学习率为0.01,最大迭代次数为4×104,动量因子为0.9。在18 400条染色体数据集的准确率为85.32%。
总体来看,基于ResNet网络的染色体分类模型的分类性能在91%以上,而基于Siamese网络的染色体分类模型性能远不如前者。原因可能是残差网络中引入的跳跃连接扩宽了网络模型的深度,使ResNet算法能比孪生网络模型学习到更多的图像细节信息,最终带来了较好的性能结果。
1.3.3 多任务学习的分类模型
多任务学习是一种推导迁移学习方法,主任务使用相关子任务的训练信号提高主任务的泛化性能,其强大的特征学习能力引起研究者关注,并将其应用于染色体分类任务中。

图16 Varifocal-Net架构图(Qin等,2019)
Zhang等人(2021)提出一种融入交错和多任务学习网络的染色体分类架构,如图17所示。该模型包含3个阶段。第1阶段,改进的HRNet(high resolution network)网络(Sun等,2019)构成的交错网络机制允许网络选择性地增强有用的特征并抑制无用的特征,甚至不仅限于高分辨率特征,从而实现特征的有效学习。第2阶段,将前一阶段学习到的高分辨率特征信息(提供的关键点信息以定位染色体)送至卷积神经子网络进行染色体联合检测,而融合剩余特征信息喂送入多层感知子网络实现染色体类型和极性的分类。最后一个阶段执行拉直染色体的任务。在收集到的32 810条染色体构成的数据集上进行测试,5折交叉验证结果显示,模型的平均分类准确率为98.1%。
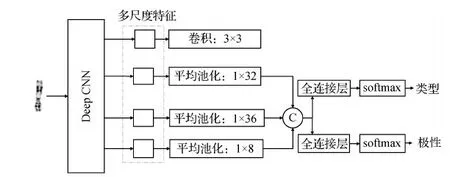
图17 多任务分类架构图(Zhang等,2021)
本文精选并阐述了近5年基于深度学习技术的染色体分类代表性算法,并按照网络设计思路介绍了模型的演变过程,总结了现有染色体分类模型相关细节信息,如表3所示。

表3 深度学习下的染色体分类代表性算法
2.1 评估使用的数据库
染色体数据库的建立过程如图18所示,主要包括中期细胞图像数据获取、染色体图像筛选和染色体实例分割获取3个阶段。

图18 染色体数据库的建立过程
2.1.1 中期细胞图像数据获取
图像数据取自广东省妇幼保健院的408名患者的6 587幅染色细胞显微图像。图像分辨率统一为1 360 × 1 024像素,96 dpi。图像文件的命名方式用于区分不同的个人ID。例如,P21000.001.A.jpg至P21000.015.A.jpg表示一个受检者为P21000个体的外周血中采集到的15幅细胞图像。每位患者至少收集8张中期细胞图片,有关隐私信息(姓名、年龄等)均已删除。
2.1.2 细胞图像筛选
经显微镜获取的中期细胞图像可能存在质量问题,如细胞杂质、噪声和拍摄模糊等,如图19所示。为此,组织16位广东省妇幼保健院经验丰富的细胞遗传学专家对低质量细胞图像开展人工剔除。最终在图像选择阶段收集到2 795幅高质量的细胞图像。
图19 带杂质细胞图像示例
2.1.3 染色体实例获取
经中期细胞图像收集和图像质量评估两个阶段后,细胞图像质量得以保证。对此,核型分析师依据第三方软件对细胞图像进行染色体实例提取,将每幅中期细胞图像分割得到46条染色体,最终形成染色体数据库。
在染色体实例提取阶段,首先从核型中分割出单个染色体实例,并将其放置在300×300像素的空白图像中心作为染色体样本。然后将图像保存到相应的文件夹中,最后由24个文件夹和126 543个染色体样本组成数据集。本文提到的临床染色体数据库是依托广东省妇女儿童医院医学遗传中心获取的临床染色体显微镜图像数据。染色体核型图像中的染色体实例由细胞遗传学专家按标签信息正确归类并调整为正确的极性方向。
2.2 参评模型及其环境配置
2.2.1 参与评估的主流模型
为了测试现有染色体分类算法之间的性能,本文在构建的临床数据集上实现并评估现有算法,从表3中精选出6种主流算法开展评估实验,如表4所示。

表4 参与评估的分类算法比较
2.2.2 实验设置
实验采用随机分层抽样将126 453条染色体组成的临床数据集分为训练数据集(80%)、验证数据集(10%)和测试数据集(10%)(Pedregosa等,2011)。参与评估的模型在相同的训练和验证数据集上训练,在同一测试数据集上评估。挑选的主流模型均在Pytorch(Paszke等,2017)框架上开发。实验环境为CPU E5-2620 v4 @ 2.1 kHz、64 GB的RAM内存和4个Nvidia GeForce RTX 2080Ti的11019MiB GPU,CentOS操作系统。
训练过程如下:首先,参评模型从ImageNet(Deng等,2009)分类任务中进行预训练和迁移。然后,利用周期性学习率(cycle learning rate,CLR)(Smith,2018)方法,在临床数据集中逐步调整各模型的表现。受限于GeForce RTX 2080Ti GPU的运行内存容量,实验的批处理大小设置为32(batch_size = 32),损失函数通过α=0.1的标记平滑(Szegedy等,2016)的交叉熵损失函数来调整,学习速率设置为10-4(lr=10-4),最大训练迭代次数的超参数设置为500。当验证损失在5个epoch没有下降时,提前终止训练过程。最后,训练结束时呈现方法的最佳训练权重恢复到相应模型。
2.3 实验结果与分析
采用准确率、精确率、召回率和F1分数作为定量评估指标。实验结果如表5所示。绝对提升是指评估性能与原有方法性能的绝对差值,相对提升是指绝对提升性能数值占原有方法性能数值的比率。具体提升如图20所示。
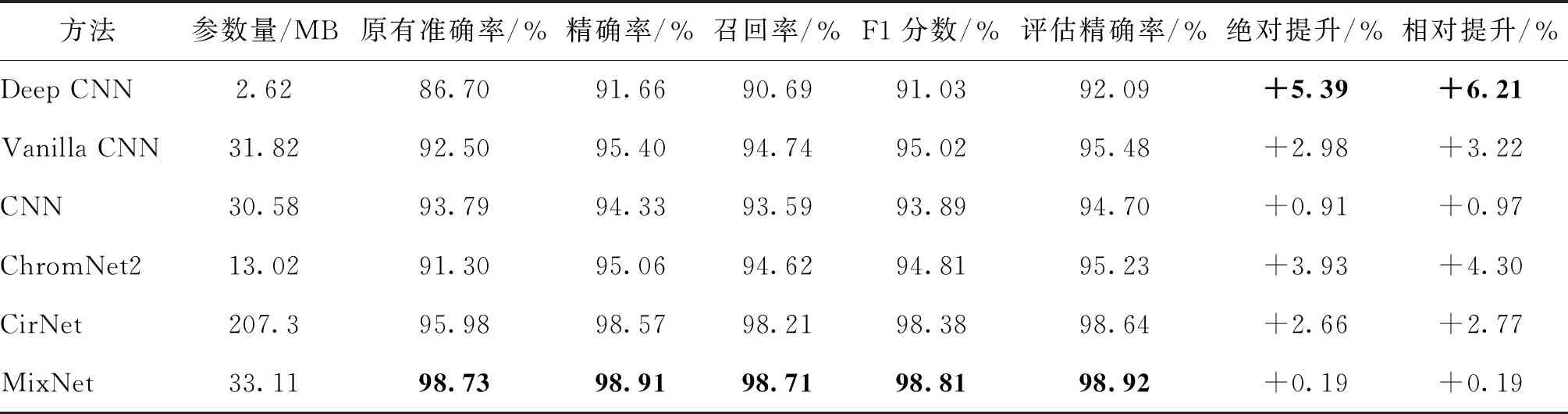
表5 参评模型在构建的数据集上的实验结果

图20 不同算法的性能提升结果
从表5可以看出,1)参数量是深度学习分类模型中的一个重要因素,参数量的改变带来的是网络分类效果的巨大变化。对染色体分类任务来说,参数量为2.62 MB的Deep CNN轻量化模型准确率为86.7%,参数量为207.3 MB的CirNet模型的准确率为95.98%,表明参数量与模型分类性能存在一定的相关性,但不包含MixNet模型。2)评价指标的不同选取方式可以客观评估模型的真实效能。参数量为33.11 MB的MixNet模型在精确率(98.91%)、准确率(98.92%)、召回率(98.71%)和F1值(98.81%)等4类分类结果上都取得了最好结果,说明MixNet模型分类稳定性较好。模型性能优劣不取决于参数规模的大小,而取决于网络结构设计过程的合理性与否。通过巧妙设计,在降低网络复杂性的同时,可以在同等条件下提升分类性能。3)所有模型均有不同程度上的性能提升。Deep CNN模型提升幅度最大。在本文选取的数据集上重新评估,Deep CNN网络的识别效果从86.7%提升至92.09%,有效缩短了与其他模型的差距。Lin等人(2020a)提出的基于改进残差神经网络的MixNet的分类精度为98.73%,有0.19%的小幅度提升。
CirNet和MixNet模型无论是初始性能还是提升后的分类效果都明显好于其他算法模型,上述两种模型的原型都是残差神经网络。运用ResNet模型开展染色体分类研究的相关算法设计可以取得不错效果,原因可能是残差连接能扩宽网络的深度和宽度,增加训练参数量,提高网络的空间复杂度,增强网络结构本身的表达能力,进而得到最为理想的分类水平。此外,大规模染色体数据量缓解了模型过拟合现象的发生。
现有方法评估后最高分类准确率为98.92%,但距高精度的临床应用仍有提升空间。因为1%的精度缺失导致的某个误诊可能使一个生命无辜放弃,因此针对网络结构的改进和大型具有代表性的临床染色体数据库的构建,是染色体分类研究在很长一段时间需要解决的重点问题。
本文总结了现有染色体分类方法难以在临床实践中广泛应用的两个关键挑战,这也是在大规模数据集下对主流模型进行性能评估的出发点。第1个挑战是目前大多数染色体分类方法的实现细节不够具体,私有数据集使不同模型之间无法进行公平比较,现有研究不可直接使用,现有方法很难复制、验证和应用。第2个挑战是当前方法是在小规模私有数据集上训练和测试的,性能不够客观。
为此,本文通过构建大规模染色体分类数据集和选取主流模型在该数据集上进行性能评估对比实验,解决了上述两个挑战。首先,详细阐述了近几年采用深度学习技术解决染色体分类任务的相关算法,介绍了模型结构、模型演变以及模型之间的联系。其次,选择6种主流染色体分类算法,将6种网络的预训练模型在126 453条染色体临床数据集上统一进行性能评估和比对实验,从不同维度分析效果提升的原因。
深度学习算法的训练需要依赖大量数据,染色体图像数据提供了染色体类别特征独特信息的成像特点,需要遗传学专家手工获取和标注,相当费时耗力。未来在基于深度学习的染色体分类研究中,不仅需要打通数据壁垒,而且需要采用数据增广技术扩大数据量,需要持续开展数据自动化标注方面的研究。随着超分辨算法的出现,基于染色体图像的重建是构建高质量临床染色体数据集的重要手段。可解释性对于以神经网络为主的方法相当重要,需要继续开展深度神经网络在染色体分类中的可解释性研究。轻量级深度神经网络如何部署到便携式医疗设备中也是一个值得研究的方向。
深度学习在医学影像中的染色体分类任务中取得了明显的研究进展,但仍需要从方法、应用等方面进一步深入研究。现有研究成果距离临床实际应用的目标还有较大差距,需要继续提高模型精度,完善结果的可解释性。
猜你喜欢染色体卷积准确率基于3D-Winograd的快速卷积算法设计及FPGA实现北京航空航天大学学报(2021年9期)2021-11-02乳腺超声检查诊断乳腺肿瘤的特异度及准确率分析健康之家(2021年19期)2021-05-23不同序列磁共振成像诊断脊柱损伤的临床准确率比较探讨医学食疗与健康(2021年27期)2021-05-132015—2017 年宁夏各天气预报参考产品质量检验分析农业科技与信息(2021年2期)2021-03-27从滤波器理解卷积电子制作(2019年11期)2019-07-04多一条X染色体,寿命会更长科学之谜(2019年3期)2019-03-28为什么男性要有一条X染色体?科学之谜(2018年8期)2018-09-29高速公路车牌识别标识站准确率验证法中国交通信息化(2018年5期)2018-08-21基于傅里叶域卷积表示的目标跟踪算法北京航空航天大学学报(2018年1期)2018-04-20能忍的人寿命长恋爱婚姻家庭·养生版(2016年9期)2016-09-07推荐访问:染色体 算法 深度