多壁碳纳米柱净化法检测茶叶中多种农药残留
来源:优秀文章 发布时间:2023-04-14 点击:
周宏霞,王妙,张梅超,张羽
威海市食品药品检验检测研究院(威海 264210)
茶叶作为天然绿色的健康饮品深受国内外消费者的喜爱。但是茶叶在种植过程中会使用一些农药,以防治和减少茶树病虫害的危害,但可能导致茶叶中残留农药而危害人体健康。因此茶叶中农残检测一直是国内外食品监管部门的监测重点。欧盟、日本等国家或地区对茶叶中农药残留做了严格要求,并制定最大残留限量(MRL),我国的GB 2763—2021《食品安全国家标准 食品中农药最大残留限量》中规定茶叶中各种农残的限量要求[1-2]。因此开展茶叶中多种农药残留的检测,对茶叶中的农残进行监控是十分必要。
农药残留前处理技术主要包括液液萃取法[3-5]、固相萃取法[6-7]、薄层色谱法和QuEChERS[8-11]。QuEChERS方法具有分析速度快、通量大、效率高且成本低等特点,能够很好地与液相色谱-串联质谱检测技术融合,满足高通量、高选择性、高灵敏度的检测技术要求。QuEChERS净化法一般采用乙二胺-N-丙基硅烷(primary secondary amine,PSA)、石墨化碳黑(graphitized carbon black,GCB)和十八烷基硅胶键合相(C18)等物质为净化剂[12]。但是茶叶基质复杂,采用一般的QuEChERS净化剂进行净化,会存在净化不彻底,存在基质干扰的现象发生。多壁碳纳米是一种新型碳纳米材料,由于具有比表面积大、吸附能力强等优点,被应用于改进QuEChERS方法的净化性能[13-15]。IC-NANO-C是多壁碳纳米柱的一种,它采用表面键合特殊官能团修饰,增加了对色素、脂肪酸等干扰物的选择性,并且通过表面去活技术,控制材料对药物的过分吸附力,保证敏感性农药的回收率。该种材料比表面积大,具有典型的层状中空结构特征,增大其比表面积,增加材料的附载能力。
近年来,用多壁碳纳米柱对茶叶进行净化,液质联用法进行检测的研究未见报道。试验采用IC-NANO-C多壁碳纳米柱对茶叶样品进行净化进行多农残留量的分析,建立一种快速、简便、高效的检测茶叶中多种农残的方法。
1.1 仪器与试剂
LC-30AD岛津高效液相色谱仪(日本岛津公司);
AB6500 Qtrap三重四极杆质谱仪(美国AB公司);
离心机(Eppendorf);
涡旋混合器(德国IKA公司);
电子天平(0.1 mg,赛多利斯);
Milli-Q超纯水器(美国Milipore公司)。
乙腈、甲酸(均为色谱纯,Thermo);
灭多威、甲拌磷、水胺硫磷、吡虫啉、毒死蜱、克百威标准物(100 μg/mL,农业部环境质量监督检验测试中心);
IC-NANO-V、IC-NANO-C(均为天津博纳艾杰尔科技有限公司)。
绿茶叶样品(购自当地市场);
空白基质样品(来自购买样品初筛后确定无残留的样品)。
1.2 试验方法
1.2.1 样品前处理
准确称取2.0 g(精确至0.01 g)制备好的茶叶粉末于50 mL离心管内,加入10 mL水涡旋混匀后,静止浸泡30 min,准确加入10 mL 1%的乙酸酸化乙腈涡旋1 min,加入4 g无水硫酸镁和1 g氯化钠剧烈震荡1 min后,按5 000 r/min离心5 min。取1 mL上清液于IC-NANO-C净化柱中,推出,反复推吸操作3~4次,上清液过0.22 μm有机滤膜,待上机检测。
1.2.2 标准溶液的配制
准确移取0.1 mL质量浓度100 μg/mL的灭多威、水胺硫磷、甲拌磷、克百威、毒死蜱、吡虫啉、氧乐果7种标准物质于10 mL容量瓶中,加入乙腈定容得质量浓度1 μg/mL的标准中间液,于-18 ℃保存。根据试验需求,按照1.2.1的样品前处理步骤处理空白茶叶样品制得空白样品基质,用空白基质溶液配制质量浓度1,5,10,20,50和100 ng/mL的标准工作溶液系列。
1.2.3 液相色谱-质谱条件
色谱柱Kinetex®C18(2.1 mm×100 mm,2.6 μm)。流动相A为0.1%甲酸水;
流动相B为甲醇;
柱温30℃;
流速0.30 mL/min;
进样量2.0 μL;
液相梯度洗脱:0~2.0 min,95% A;
2.0~9.0 min,95%~5% A;
9.0~12.0 min,5% A;
12.0~12.1 min,5%~95% A;
12.1~14.0 min,95% A。
离子源采用电喷雾离子源(ESI)正离子模式;
扫描模式采用多反应监测(MRM)模式;
离子源温度500 ℃;
电喷雾电压5 500 V;
气帘气压35 psi;
雾化气压55.0 psi;
辅助气压55.0 psi。7种农药的质谱条件见表1。

表1 MRM条件下的7种农药的质谱参数
2.1 仪器条件的优化
2.1.1 质谱条件的优化
将各农药配制成50 μg/L的混合标准溶液。直接注入质谱仪,采用正离子扫描模式,得到准确的母离子,利用质谱仪自动优化功能,筛选二级碎片离子信息,获得碎片离子及碰撞能量,并将母离子和2个信号较强的子离子组成监测离子对,以MRM多反应监测模式进行检测。
2.1.2 液相条件的优化
试验比较甲醇-水、乙腈-水、乙腈-0.1%甲酸水溶液、甲醇-0.1%甲酸水溶液、作为流动相时对化合物分析结果的影响。结果表明,采用甲醇-0.1%甲酸水的流动相,7种物质分离度好,相应值也高,因此采用此流动相。在该条件下所得的多反应监测总离子流色谱图见图1。
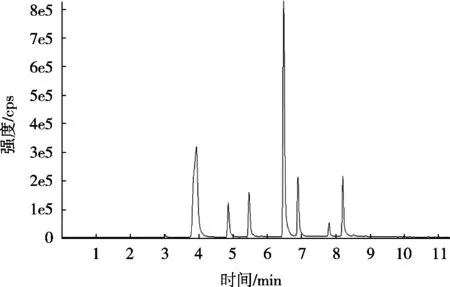
图1 7种农药的总离子流图
2.2 样品处理条件的优化
2.2.1 净化柱的选择
采用IC-NANO-C柱、IC-NANO-V柱及PSA净化管进行比较,结果发现IC-NANO-C净化效果最好,回收率比较稳定,提取液颜色浅,适合于茶叶这类高色素,复杂基质样品的净化。
2.2.2 提取液的选择
在农药残检测过程中,常用的有机溶剂有乙腈、甲醇、乙酸乙酯等,这几种提取剂的提取效果相差不大。但是由于茶叶的基质比较复杂,甲醇、乙酸乙酯作为提取剂时,得到的提取液颜色较深,不利于后续的净化,而乙腈作为提取剂时其提取出的咖啡因、色素等杂质相对较少,且能同时沉淀蛋白等,因此选用乙腈,并且这7种物质为正离子模式电离,在酸性条件下响应值较高,因此采用1%乙酸酸化乙腈进行提取。
2.3 基质效应
在液质检测过程中基质效应会影响方法的灵敏度、精密度和准确度。试验运用相对响应值的方法[16]评价7种农药的基质效应。
式中:A为基质匹配标准溶液的响应值;
B为乙腈溶剂标准溶液的响应值。
当ME为正值是基质增强,为负值时是基质抑制。由表2可知,7种茶叶中灭多威、水胺硫磷、甲拌磷、克百威、吡虫啉、氧乐果的ME均为负值,毒死蜱的ME为正值。茶叶对这7种物质都有一定的基质效应,因此采用空白基质提取液配制标准溶液,进行外标法定量。
2.4 方法的线性及检出限
按照1.2.2配制系列质量浓度的混合标准工作溶液,按照1.2.3的仪器条件进行测定,以质量浓度为横坐标,7种待测农药的峰面积为纵坐标建立校准曲线。采用空白基质样品加标的试验来确定方法的检出限,以定量离子对和定性离子对的3倍信噪比(S/N)确定方法的检出限。由表2可知,7种待测农药均在1~100 ng/mL范围内线性良好,相关系数(r)均不小于0.996 2,SLOD为2.0~5.0 μg/kg。

表2 7种农药的线性方程、相关系数、检出限和基质效应
2.5 方法的精密度及回收率
基质效应对样品检测具有一定影响,为提高检测的准确性,采用空白绿茶样品进行加标试验。取空白的茶叶样品,对7种农药进行5,10和50 μg/kg 3个水平的加标回收试验,每个浓度做6个平行样品,按照1.2的试验方法进行试验,结果如3表所示。结果显示,7种农残的平均回收率为78.0%~100.9%,相对标准偏差为1.17%~6.22%,满足农残检测的要求。

表3 方法回收率和相对标准偏差
试验采用多壁碳纳米柱对茶叶复杂基质进行进化,结合液质联用仪进行快速检测。IC-NANO-C柱净化效果好,能去除茶叶中的色素干扰,农药残留回收率高。液质联用质谱仪能对样品进行快速定性、定量,解决同时检测茶叶中多农残的检测问题。通过试验发现:7种农药的SLOD为2~5 μg/kg,回收率为78.0%~100.9%,该方法简单、快速、准确,适合用于茶叶中多农残的检测。
猜你喜欢乙腈净化回收率高纯乙腈提纯精制工艺节能优化方案煤化工(2022年3期)2022-07-08不同形态氮肥对棉花15N回收率和产量的影响中国土壤与肥料(2021年5期)2021-12-02吹扫捕集-气相色谱-三重四极杆质谱法同时测定饮用出厂水中6种卤乙腈色谱(2021年7期)2021-06-07全国农膜回收率年底前达到80%以上今日农业(2020年22期)2020-12-14这条鱼供不应求!虾蟹养殖户、垂钓者的最爱,不用投喂,还能净化水质当代水产(2019年3期)2019-05-14肌肤净化大扫除STARTCoco薇(2017年7期)2017-07-21陶色净化金色年华(2016年23期)2016-06-15丁二酮肟重量法测定双乙腈二氯化中钯的含量中国资源综合利用(2016年10期)2016-01-22提高梅山钢铁企业生活区电费回收率的研究实践电力需求侧管理(2014年4期)2014-03-20给未成年人净化出一片晴朗的天空中国火炬(2012年11期)2012-07-24推荐访问:纳米 农药 残留