4例糖原贮积症IXa型报道及文献回顾*
来源:优秀文章 发布时间:2022-12-04 点击:
陈思杏 ,吕自力 ,唐 清 ,陆霜霜 ,黄 丽 ,陈秀奇 ,兰连成,单庆文△
(1.广西医科大学第一附属医院,南宁 530021;
2.右江民族医学院附属医院,百色 533000)
糖原贮积症IX 型(glycogen storage disease type ixa,GSD IX)是磷酸化酶激酶(PHK)缺陷所致的先天代谢性疾病,发病率约为1/10万[1]。GSD IXa型是由编码PHK 的α 亚基的PHKA2 基因突变引起的,以X连锁隐性的方式遗传,是GSD IX型最常见的类型。PHK由4个不同的亚基组成,分别为α、β、γ 和δ 亚基。其中α 调节亚基是由位于X 染色体的PHKA2基因编码。PHKA2基因突变可能会损害亚基、PHK 全酶的稳定性或导致酶的失调[2]。本文收集了4 例GSD IXa 型患儿的临床资料,并结合文献进行了比较和分析,以加深对该病的认识。
1.1 研究对象 通过检索2016—2022 年我院住院电子病历系统,筛查出最终诊断为GSD IX,同时为PHKA2基因突变的患儿4例。
1.2 方法 收集患儿的病例信息,包括一般资料、临床表现、辅助检查、诊治及随访。在获得患儿监护人知情同意的情况下,对患儿进行肝组织穿刺活检或基因检测,并收集相关结果。
2.1 一般资料 4 例患儿均为男孩,发病年龄在10~42 个月,因“肝功能异常”入院,其中2 例患儿是在入园体检发现肝功能异常。例1 患儿外祖父有“肝癌”病史,曾祖父因“肝硬化”去世。
2.2 临床表现 4 例患儿均存在肝肿大,无脾脏肿大。肝脏肿大程度可分为:轻度:肝肋下3 cm以内;
中度:肝肋下3 cm 以上至脐;
重度:肝脏超过脐水平;
极重度:肝多已入骨盆,并横过中线。按照上述标准,4 例患儿肝肿大程度均为中度肿大,质地中等,边界钝。例1患儿有长期腹胀,入院查体腹部膨隆;
例2 患儿伴有上呼吸道感染症状,表现为咳嗽、流涕。例1、例2 患儿身材矮小(<P3),例3 患儿身材中等(P25~P50),例4患儿身材偏矮(P10~P25)。
2.3 实验室及影像检查 病程中4 例患儿均有肝功能异常,表现为转氨酶升高,例2患儿不伴谷氨酰转肽酶升高。2例患儿胆汁酸升高。例1、例2患儿存在轻度贫血,其中例2 患儿为小细胞低色素性贫血,符合既往地中海贫血的诊断。在代谢指标方面,3 例患儿有高乳酸血症;
2 例患儿有高甘油三酯血症;
2 例患儿空腹血糖偏低;
例1 患儿血串联质谱发现甲硫氨酸降低。所有患儿的血铜、铜蓝蛋白、血氨未见异常,未见眼K-F环。4例患儿腹部B超示肝脏肿大伴或不伴肝实质回声增粗增强,其中3 例患儿行无创肝纤维化超声检查,肝脏硬度值未见异常,但其中1例患儿提示存在轻度脂肪肝,见表1。

表1 4例GSD IXa患儿临床特征
2.4 病理检查 4 例患儿均进行了肝组织活检,均符合糖原贮积症。光镜下可见肝小叶结构存在,肝细胞体积普遍增大,其中例3 患儿肝细胞呈透明样改变。3 例患儿汇管区增宽,见少许纤维组织增生。3例患儿马松染色显示胶原纤维增生;
4例患儿过碘酸希夫染色阳性(PAS:+~+++),胞质内可见特征性红色颗粒,经淀粉酶处理后,红色颗粒消失。例1~例3患儿病理肝炎纤维化评分为G1S1,例4患儿评分为G0S0。例1患儿的肝脏病理组织学改变情况,见图1。

图1 例1患儿肝脏组织穿刺病理图片(×200)
2.5 基因分析 行一代基因测序,4 例患儿均检测到PHKA2基因突变,遗传来自母亲。例1患儿在第33位外显子检测到c.3570dupA移码突变,该变异为新突变,会使所编码的蛋白质自第1 191 位氨基酸Asp 开始发生编码紊乱,并使得蛋白质翻译提前终止。例2 患儿在第25 位外显子检测到c.2746C>T(p.R916W)错义突变,该突变既往已有报道[3],在2746号核苷酸由胞嘧啶C变为胸腺嘧啶T的半合子突变,导致第916 位氨基酸由精氨酸变成色氨酸。例3 患儿为exon27-33 半合子缺失,大片段缺失,属于低频变异,为致病突变,该突变在国外亦有相关报道[4]。例4 患儿在第5 位外显子检测到c.496C>T,该突变为无义突变,使得编码蛋白质发生截短而丧失正常功能。该突变未见文献报道,ESP6500、千人基因组、dbSNP 和ClinVar 数据库未见收录,该变异亦为新突变。
2.6 治疗与随访 住院期间予复方甘草酸苷、熊去氧胆酸护肝利胆治疗,4 例患儿肝转氨酶较入院时降低。针对原发病的治疗,其中3 例患儿使用规律服用生玉米淀粉,另外1 例患儿暂予高热卡奶粉喂养,当时患儿并无低血糖的发生。4 例患儿均在随访当中,肝功能、乳酸逐渐恢复正常,其中例4 患儿随访时长6 年,患儿的肝功能好转,空腹血糖、乳酸正常或偏低,身高、体重也随年龄增长,虽生化指标好转,但肝脏肿大仍持续存在。
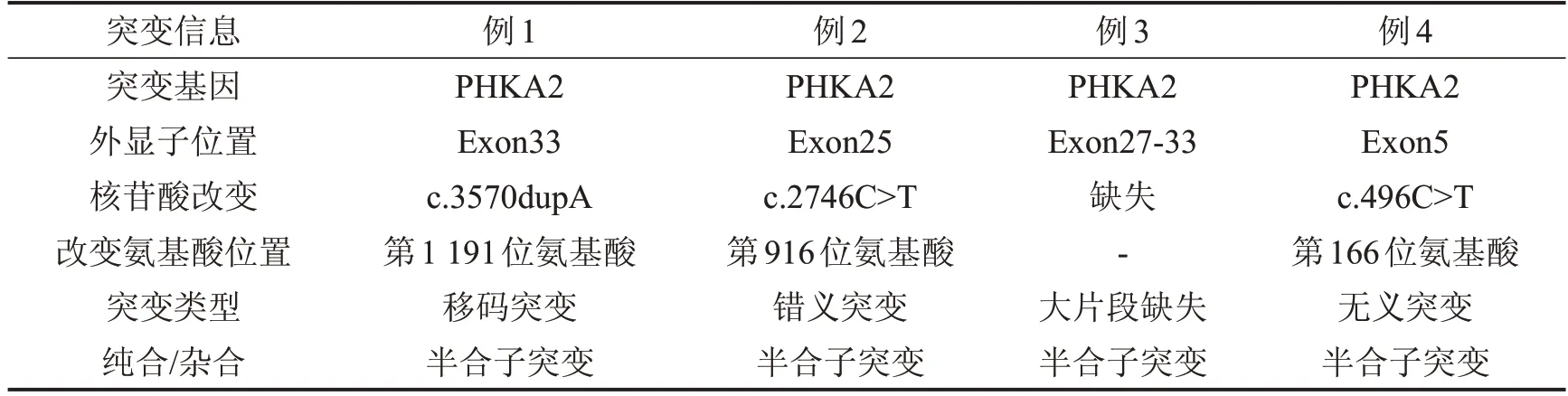
表2 4例GSD IXa患儿基因突变信息
磷酸化酶是糖原分解的关键酶,催化糖原生成葡萄糖-1-磷酸,而磷酸化酶需要PHK的激活。PHK活性减少或失活可直接影响糖原的分解,造成糖原沉积。GSD IXa 型又称为X 连锁肝糖原增多症(XLG)。XLG 患者可分为两种亚型:其中在XLG 1型中血和肝脏的PHK活性降低,而在XLG 2型中血细胞的PHK 活性正常,不同患者的肝脏PHK 酶活性可能不同[2]。XLG 2 型是由于PHKA2 基因突变,引起α 亚基的一级结构改变,临床表现通常比XLG 1型更轻微[5]。PHKA2基因位于染色体XP22.13,其包括33 个外显子,并跨越65 kb 及更多的片段。GSD IXa 型是X 连锁隐性遗传的疾病,本文4 例患儿均为男孩,既往研究大部分患者为男性,但亦有GSD IXa 型是女性患者的报道[6-8],PHK 活性的降低,这可能与其中X染色体失活有关。
回顾文献,GSD IXa 型患儿确诊年龄多为幼儿期,年龄最小为新生儿期发病。该病有各种临床表现,多数为轻型,包括肝脏肿大、肝功能异常、生长迟缓、语言发育迟缓、低血糖、高乳酸、高甘油三酯、间断腹泻或便血、特应性皮炎、肌张力减低[7,9],但也可出现因低血糖而引起的抽搐、乳酸酸中毒继发的肾小管酸中毒[9-10]、肝硬化[11-13],甚至肝腺瘤[14]等严重表现。一项荟萃分析了183 例GSD IXα 型患儿信息,有15个家庭有GSD IXα家族史。在被详细报道的患儿中,存在肝肿大占93.2%(164/176),出现空腹低血糖者占43.8%(53/121),伴有高甘油三酯血症者占65.3%(64/98)[15]。本研究中的患儿临床表现与先前研究类似,4 例患儿均有肝功能异常及肝脏肿大,3 例患儿(例1~例3)有高乳酸血症,2 例甘油三酯升高,2例患儿有生长发育迟缓。病程中曾有2例患儿患儿空腹血糖偏低,在禁食过程中未发生低血糖症状,这与GSD IXa 型糖异生途径完整,弥补了糖原分解途径障碍的不足有关。临床中GSD IXa型可能出现血糖正常的酮血症,故建议有条件者应同时检测血酮及尿酮。严重低血糖影响三羧酸循环,谷氨酸和天冬氨酸增加导致神经元细胞肿胀或死亡,或直接造成海马体的体积减少和神经元突触改变,临床表现为低血糖性痫性发作、认识和语言发育延迟、周围感觉神经改变。与GSD I型相比,GSD IXa 型神经系统表现多轻微,因为后者较少出现严重的低血糖和乳酸酸中毒[10]。本研究中,4 例患儿均未出现神经系统表现,考虑可能由于患儿无严重低血糖,未对神经系统造成明显影响。因此,关于GSD IXa 型的其他表现仍需要进一步观察及更多的临床研究。
临床上,表现为不明原因的肝肿大、肝功能异常伴代谢指标异常的患儿,考虑存在糖原贮积症可能时,可完善肝穿病理检查协助诊断。GSD IXa 型肝脏组织病理炎症程度多为G1~G2,纤维化程度多数为S1~S3,少部分为S4。其病理表现多样,包括肝细胞脂肪变性及肿胀、肝细胞胞质结构消失、小叶间隔形成、汇管区纤维化、糖原沉积,严重者有门脉三期纤维化、结节性硬化。在本研究中,4例患儿病理均有肝细胞肿胀、糖原沉积,无脂肪变性、肝硬化表现,其中3 例患儿纤维化程度为1 级,1 例患儿无纤维化。例4 患儿转氨酶较例1、例2 患儿高,病理无肝炎纤维化,表明生化异常不能完全预测肝损伤,这与既往报道一致[16]。糖原贮积症的病理组织学可能与分子诊断并不一致,如部分病理符合GSDⅢ型的患儿基因诊断可能为GSD IXa、GSD IXc、GSD Ⅵ型;
而病理表现为GSD Ⅳ型患儿,基因诊断符合GSD IXa或GSD Ⅵ型[14]。因此,如有条件应进一步行基因检测以明确诊断。
目前,研究发现PHKA2 基因有多种突变类型。截至2022年4月,人类基因突变数据库(the human gene mutation database,HGMD)记录了166 个PHKA2基因突变。目前发现错义突变/无义突变85个、小片段缺失32 个、剪切突变17 个、小片段插入16个、大片段缺失12个、小片段插入缺失2个,大片段插入2 个,以错义突变为主,尚未发现重复突变、调节突变及复杂突变。本研究4例患儿均为半合子突变,移码突变、错义突变、无义突变、大片段缺失各有1例。回顾文献,与错义突变相比,移码突变对蛋白质的正常功能影响更大。PHKA2 包含可能参与PHK 调节的假定磷酸化位点和钙调蛋白结合位点,基因突变影响PHK 中a 亚基的稳定性,导致PHK 活性降低[17]。除韩国外,亚洲其他国家多为错义突变、无义突变或移码突变。韩国人群的PKKA2基因突变谱中多为大片段缺失,法国及日本则少见[4,17-18],目前国内尚无PHKA2基因大片段缺失的相关报道。本研究例3 患儿检测到PHKA2 基因外显子27-33半合子大片段缺失,临床表现为肝脏肿大、肝转氨酶升高、高乳酸血症和空腹血糖偏低,这与既往韩国人群中报道的该种突变表现一致。而法国人群中发现有内含子26-33 的缺失,其临床表现在文献中并未详细描述[18]。本研究中例1患儿的第33 位外显子c.3570dupA 及例4 患儿的第5 位外显子c.496C>T 为致病性新突变,在ESP600、千人基因组、dbSNP 数据库未见收录,增加了PHKA2 基因突变种类。
此外,还发现GSD IXa 型的生物化学及临床表现具有高度变异性,轻者仅有无症状肝肿大,重者发生肝硬化或癫痫样抽搐,其是否为良性病变仍存在一定的争议。因此,该病较GSD IX 型的其他亚型诊断更为困难。既往研究显示,GSD IXa、GSDIXb、GSD IXc 型的平均诊断年龄分别为4 岁、2.34岁、1.8 岁[15]。GSD IXa 型患儿随着年龄增长,GSDIXa 型对葡萄糖的需求减少,因低血糖所致症状显得更轻微,具有独特的生长模式,即早期生长发育迟缓,后期可追赶达同龄儿水平[15,19],积极的生玉米淀粉治疗可改善患儿低血糖症状、身材矮小及所致的心理困扰,甚至可改善肝纤维化、肝硬化[11]。本研究中,4 例患儿予生玉米淀粉喂养或高热量的食物喂养,例1、例3患儿随访时间尚未超过半年,身高与入院时处于同一百分位。例4 患儿随访6 年,身高水平已达P50,定期检查转氨酶较前好转(末次ALT 27U/L,AST 11U/L),仍有轻度肝大(肋下2 cm)。与既往报道类似,该病患儿生化、代谢指标改善,肝脏肿大仍可较长时间存在。国外指南建议注意定期随访(3~12 个月),监测肝功能、凝血指标等,每1~2年行无创肝纤维化超声检查明确有无肝硬化或腺瘤[20]。
综上,有肝大及代谢指标异常的男性患儿需要考虑GSD IXa 型的可能,GSD IX 型是一种糖原分解异常,糖异生途径完整的糖代谢障碍疾病。GSDIXa型的临床表现多样,与之相关的PHKA2基因变异谱广泛,诊断时间可能相对其他亚型较长,基因检测可早期诊断该疾病。在明确诊断GSD IXa 型后,建议早期积极治疗并长期随访,进行临床症状和实验室生化指标的评估,对该疾病患儿进行优化管理。
猜你喜欢 糖原基因突变纤维化 线粒体自噬在纤维化疾病中作用的研究进展中华实用诊断与治疗杂志(2022年1期)2022-08-31炎性及心肌纤维化相关标志物在心力衰竭中的研究进展中国现代医生(2022年21期)2022-08-22恩替卡韦联合安络化纤丸治疗慢性乙型肝炎肝纤维化的研究中国药学药品知识仓库(2022年9期)2022-05-23肝纤维化防治面临的挑战家庭医学·下半月(2022年3期)2022-04-07携带线粒体12S rRNA基因突变的新生儿母系家族史分析中国听力语言康复科学杂志(2021年6期)2021-12-21长期服用奥氮平对大鼠糖原代谢及糖转运蛋白表达的影响中国医药导报(2019年3期)2019-03-18“基因突变和基因重组”复习导航中学生理科应试(2017年6期)2017-09-27抑制糖原合成激酶3a可减轻6—羟基多巴引起的SH—SY5Y细胞损伤现代养生·下半月(2016年6期)2016-10-21什么是糖原异生求医问药(2009年7期)2009-08-31浅议糖原的分布及肌糖原的利用和转变中学生物学(2009年1期)2009-02-23推荐访问:糖原 文献 回顾