融合多模态数据的药物合成反应的虚拟筛选
来源:优秀文章 发布时间:2023-04-14 点击:
孙晓飞,朱静远,陈 斌,游恒志*
(1.中国科学院 成都计算机应用研究所,成都 610041;
2.中国科学院大学 计算机科学与技术学院,北京 100049;
3.哈尔滨工业大学(深圳)理学院,广东 深圳 518055;
4.哈尔滨工业大学(深圳)人工智能研究院,广东 深圳 518055)
药物合成中的有机化学反应的发现依赖于实践经验和化学机理支配的“化学直觉”,实验人员试图定性地识别有机化学反应中的模式,以确定反应产物和反应效率。然而,这种方法受到了很多因素的限制,包括反应的复杂性、活性悬崖、对机理理解的缺乏以及人工处理大数据的艰难。基于计算机的虚拟筛选[1-5]已经成为吸引化学家的重要方案,主要因为它不需要机理的理解,化合物结构可以用分子性质的数值表示来表征,从而量化数以千计的候选分子的化学性质。在实验和文献数据的基础上,虚拟筛选可以通过计算机模型来对药物合成反应的结果和催化剂的选择性进行量化。
机器学习在化学领域已成功应用于药物虚拟筛选[3-5]、分子生成[6]、有机反应预测[7-8]、催化剂筛选[9-10]、材料发现[11]、计算机辅助合成设计[4,12]和反应条件优化。线性回归是传统的反应预测和分析工具[13-14],它假设反应物的物理特征和反应性之间存在线性关系,可以根据反应的机制假设人工对输入变量进行选择,所以非常符合数据科学家的思维和统计方式。Hammett[15]在线性自由能关系的推断中使用线性回归拟合化合物描述符和输出是一个代表性工作。长期以来,由于分子特征的多维性和反应空间的复杂性,很难生成足够完整和一致的数据,从而限制了机器学习的发展[11]。如今,高通量实验(High Throughput Experimentation,HTE)已经成为逐步扫除这一障碍的有效手段[8,16-20]。Ahneman 等[21]使用了支持向量机(Support Vector Machine,SVM)和随机森林(Random Forest,RF)等方法在4 000 多个高通量实验数据中预测了Buchwald-Hartwig 偶联反应的产率。此外,Zahrt等[22]通过RF 对1 000 多个反应中的手性磷酸(Chiral Phosphoric Acid,CPA)催化剂的对映选择性进行了预测。
使用计算机对化学反应进行虚拟筛选的流程如图1 所示,首先从已有的化学反应数据库和文献中提取分子的简化分子线性输入(Simplified Molecular-Input Line-Entry System,SMILES)或分子指纹,或者使用Gaussian 等密度泛函(Density Functional Theory,DFT)工具对这些分子进行结构优化并计算出与反应有关的性质;
然后,用这些物理和化学性质构建出分子描述符,再选用合适的机器学习方法进行建模;
最后对数据集中待分析的反应进行筛选。这种方法对于数据科学家来说直观有效,不需要关注反应机理的理解,已经成为化学合成预测的标准流程。该流程的成功与否,取决于两个关键因素:1)所选的DFT 特征或者分子指纹,以及用它们构建的描述符是否准确;
2)机器学习方法是否有效。药物合成相关的有机反应预测经过几十年发展,在这两个方面仍受制约。下面对这两个问题进行详细描述:

图1 对药物合成反应进行虚拟筛选的流程Fig.1 Flow of virtual screening of drug synthesis reactions
1)对于基于量子力学的DFT 特征来说,针对不同反应进行特征的选择一直是药物合成相关的预测需要面对的难题,特别是对反应产率和选择性预测的特征选择往往有很大差别。若能降低特征选择的难度,将为药物合成相关的反应预测带来促进作用。对于使用SMILES 和分子指纹的序列特征来说,对于三维(3D)结构信息表达不足是一直存在的困难。这是由SMILES 作为一种简化的分子结构线性表示以及分子指纹的算法本质决定的。
2)在药物合成相关的化学反应预测中,传统的机器学习方法如SVM 和RF 等,甚至是线性回归方法一直占据主流。由于“维度灾难”的存在,随着特征维度的上升,所需的化学反应数据急剧上升,大幅超出了人工实验的工作量。而高通量实验的出现,使这一问题逐步得到缓解。如今,如何将高通量化学反应数据应用于深度学习,已经摆在数据科学家面前。然而,由于对化学知识的缺乏和反应数据相对稀少(相对于传统深度学习应用领域,如图像、视频、音频和文本),深度学习方法在这一领域的研究仍然罕见。虽然已经有一些工作对文献中的反应数据提取SMILES 后使用深度学习方法进行预测[23],但如何使用深度学习方法对日渐累积的DFT反应数据进行虚拟筛选仍亟待研究[4,24]。
针对上述问题,本文主要工作如下:1)提出基于加权平均占位和分子DFT 性质的描述符,并用于4-甲基苯胺与芳基卤化物的C-N 偶联反应的预测。这种描述符以构象能量计算权重,得出多个构象的平均值来近似反应发生时的构象概率分布。2)提出一种针对DFT 计算性质的图卷积网络,用量子力学(Quantum Mechanics,QM)计算的原子性质作为图节点的输入特征,并融合分子指纹特征进行预测。使用这种网络在CPA 催化的N,S-缩醛反应上进行验证。据了解,现在尚无这种使用DFT 计算的性质构建的图卷积网络,这是一种将QM 特征引入深度学习方向的有益尝试。3)针对现有工作对多模态数据的表示和利用不足,采用基于卷积神经网络(Convolutional Neural Network,CNN)或图卷积神经网络(Graph Convolutional Network,GCN)[25]等深度学习手段,将所提出的3D描述符与其他来源的分子描述符融合起来,应用于反应产率和对映选择性的预测中。
在C-N 偶联反应产率预测中,采用格点精度为1 埃(0.1 nm)的加权平均占位描述符,相较于文献[21]和文献[18]提高了3和2个百分点,对仅使用DFT计算的描述符有明显优势。在N,S-缩醛反应的对映选择性预测中,在随机划分的数据集上误差从文献[22]的0.152降到0.147,验证了方法的有效性。
1.1 三维分子描述符
比较分子场分析(Comparative Molecular Field Analysis,CoMFA)作为一种使用了三维空间信息的定量构效关系(Quantitative Structure-Activity Relationship,QSAR)方法,在不对称催化相关问题上已有近20年的历史。该方法用分子力学力场近似范德华相互作用,用库仑势确定静电相互作用。这种分子描述符也考虑了分子的三维结构,被认为具有探索化学空间的潜力。该方法被首次用于分析环戊二烯和3-乙烯基恶唑烷-2-酮的Diels-Alder反应中含有膦恶唑啉或双恶唑啉配体的催化剂[26]的对映选择性,以及在PhCHO 中添加Et2Zn[27]时氨基醇催化剂的对映选择性。后来的工作采用了类似的基于CoMFA 的方法,结合了半经验[28]和量子力学相互作用能,并用于不同的不对称催化反应,如氧烯丙基阳离子[29]环加成反应和手性sparteine 替代物的不对称锂化反应[30]。在最近的杰出工作中,Zahrt等[22]介绍了一种与CoMFA完全不同的新方法,该方法关注催化剂分子的众多构象,并将构象的平均空间占有率(Average Steric Occupancy,ASO)作为描述符。
1.2 药物合成反应筛选中的多模态数据融合
随着高通量实验和机器学习在药物合成反应预测中的应用和发展,不同来源、类型和分布的多模态反应数据被产生出来,每种数据都包含特定的信息,如SMILES、分子指纹、量子力学性质和各种人工设计的2D 和3D 描述符。药物合成反应筛选中的多模态数据具有维度差异大、信息容量差异大和信息类别多样的特点。由于不同模态数据的维度和物理意义不同,对它们的利用非常困难。现有的工作都使用单一模态的反应数据,如Ahneman等[21]使用量子力学计算的分子、原子和振动描述符,采用CoMFA 的一系列研究使用分子力学力场近似的相互作用力为基础构建的描述符。
多模态融合负责联合多个模态的数据进行分类或者回归,在深度学习的很多领域应用广泛。它还有其他常见的别名,如多传感器融合和多源信息融合。单模态的机器学习将信息表示为计算机可以处理的数值向量并进一步抽象为更高层的特征向量,而多模态机器学习可以通过利用多模态数据之间的互补性,学习到更好的特征表示,如图2 所示。常见的机器学习模型都可以用于多模态融合[31]。
图2 多模态数据融合Fig.2 Multimodal data fusion
1.3 图卷积神经网络
可以使用图卷积神经网络来建模分子相关的问题,将一个化合物构建为一个图形,它的节点代表原子,连接它们的边代表键。在图卷积(图3(a))中,对于一个节点,将特征和邻居馈送到两个密集层中,然后添加密集层的输出作为节点的新特征。对具有相同度的节点计算新特征时共享权重。在一个化合物中,如果一个原子a总共有n个邻居,那么它经过图卷积后的新特征可以表示为:
其中:Wa为节点a的权重;
Wr是相邻节点的权重;
b是偏差,σ是激活函数。从节点a出发的箭头表示原子a及其相邻原子的密集层,权重分别为Wa和Wr。
与CNN 中的池化层类似,图池化层(图3(b))也被用于给化合物分子编码。图池化是返回原子及其邻居中最大或平均特征的操作,可以在不增加其他参数的情况下增加感受野:
经过图卷积和图池化,每个原子都有一个特征向量;
但为了进行最终预测,需要为整个分子图提供一个固定大小的特征向量。图聚集层(图3(c))将化合物分子中所有原子的特征向量相加,以获得分子的特征向量:
将这三种操作按图3 中黑色箭头所示的顺序进行组合,就可以构成不对称反应催化剂筛选网络中的图卷积网络模块。

图3 图卷积神经网络Fig.3 Graph convolution neural network
本文提出了一种基于加权平均占位的3D描述符,它在三维空间中整合分子级别性质和原子级别性质,为机器学习模型提供良好的输入特征,并将这种方法用于钯催化的Buchwald-Hartwig偶联反应的筛选;
还提出一种基于图卷积的多模态模型,通过将量子力学性质和分子指纹融合起来来筛选N,S-缩醛反应中的手性磷酸催化剂。下面介绍分子描述符合网络的结构的一些实现细节。
2.1 基于Boltzmann分布的加权平均占位描述符
在特征选择和模型建立过程中,需要考虑描述符对有机反应的适应性,即要寻找一组不受限于特定机理假设的描述符,以区分和描述反应之间的差异。基于这种考虑,在Buchwald-Hartwig 偶联反应的产率预测中引入一种3D 描述符(图4(a)),它首先根据结构优化后的分子3D坐标构建出一个三维网格,将分子置于网格中心并采用自开发的分子对齐算法进行对齐,然后统计每个网格点上原子的量子力学性质。考虑到单一的构象不能很好地表示反应发生时构象的复杂情况,使用Boltzmann 分布计算不同构象所占比例,生成加权平均的3D构象描述符。
其中:p是构象所占比例,k、i是构象的序号,E是构象的能量,R是理想气体常数,T是开尔文温度。di是单个构象的描述符,d是经过加权计算后的分子描述符。
3D 坐标不仅表示原子的空间信息,而且对描述符中其他物理性质起到明确的位置编码作用。其中的分子和原子性质是由Gaussian 计算得到,如最高占据分子轨道(Highest Occupied Molecular Orbital,HOMO)、最低占据分子轨道(Lowest Occupied Molecular Orbital,LUMO)、偶极矩、容积、密立根电荷等(图4(a))。这些性质的都是易于计算的且经过初步筛选以确保它们的有效性和通用性。

图4 基于加权平均占位的3D描述符及CNN特征融合模型Fig.4 3D descriptor based on weighted average occupancy and CNN feature fusion model
2.2 反应产率筛选网络架构
融合多种来源的分子特征数据,对药物合成相关的有机反应进行筛选,是本次工作的重要目标。基于加权平均占位的3D 描述符可以将原子级别的量子力学特征与立体空间特征融合起来。分子整体的量子力学性质如HOMO、LOMO 等也是与反应机理相关的重要特征,在3D描述符中并不能很好表达这种特征,在此将多个分子级别特征表示为一维向量描述符。3D 描述符数据的维度巨大,采用卷积操作构建网络以减少过拟合,而对于分子级别特征直接使用密集层构建网络,然后将两种特征进行融合(图4(b))。损失函数的形式如下:
其中:x是样本,y是化学实验得到的观察值。
2.3 不对称反应中催化剂筛选的网络架构
使用DFT 计算的量子力学性质具有化学意义且精度高,但由于DFT 计算及其数据的复杂性,至今未见到有工作将量子力学性质用图卷积的方式来表示和建模。这里从Gaussian的优化结果中提取每个原子的3D 坐标,使用RDKit 提取分子的邻接矩阵以构建图结构,并将DFT 计算的量子力学性质作为每个图节点的属性向量。这样构建起来的图卷积网络,接受的原子性质和提取的特征具有化学意义,符合化学家的化学直觉。接受SMILES 或分子指纹作为输入的图卷积网络已经被应用于药物虚拟筛选中[25,32]。构建一个网络将这些不同种类的特征融合起来,可以发挥优势互补的作用(图5)。将这个模型应用于手性磷酸催化的N,S-缩醛反应。

图5 基于图卷积的量子力学特征与分子指纹融合模型Fig.5 Quantum mechanical feature and molecular fingerprint fusion model based on graph convolution
3.1 有机反应数据和评价指标
本文在两个药物相关的有机合成反应上进行了实验,所采用的数据划分方法与文献[21]和文献[22]等保持一致,以便于比较。第一个数据集是Buchwald-Hartwig 偶联反应,该反应的底物含有杂原子-杂原子键的五元杂环(例如异恶唑),这些杂环化合物具有药物样特征,但被成功筛选为候选药物的数量仍然不足[33]。因此,使用人工智能(Artificial Intelligence,AI)预测异恶唑存在下Buchwald-Hartwig 反应的性能是很有必要的。该反应数据集包含3 960 个高通量实验的反应物、催化剂、添加剂、溶剂、产物和产率。将反应按添加剂分成4 个测试集,从而可以测试添加剂对反应带来的影响(表1)。

表1 Buchwald-Hartwig反应的数据集划分Tab.1 Splitting of Buchwald-Hartwig reaction dataset
第二个虚拟筛选数据集是手性磷酸催化的N,S-缩醛反应。该数据集中反应数量稀少,且该数据集的任务之一是探究底物和催化剂在不对称催化反应预测中的重要性。因此在数据划分时,首先将催化剂和底物按训练用途和测试用途进行划分,然后将它们组合以形成1 个训练集和3 个测试集(表2)。实验结果证明这种划分可以反映出不同模型对催化剂和底物在反应预测中的表达能力的高低。

表2 N,S-缩醛反应的数据集划分Tab.2 Splitting of N,S-acetal formation dataset
评价指标采用与已报道工作相同的R2(R-squared)和RMSE(Root Mean Square Error),其中R2指标计算方式为:
其中:分子部分表示观察值y与预测值的平方差之和;
分母部分表示观察值y与均值的平方差之和。
3.2 实验细节
在对药物合成相关反应的数据进行处理时,出于数据一致性的考虑,对于分子统一使用了Gaussian 计算出所需要的性质。为了减少人工数据分析,开发了自动化软件将分子提交给Gaussian 进行计算,并提取和解析结果。在计算催化剂的性质时,使用ChemDraw、Gaussian 创建初始的分子结构,或者从Cambridge Structural Database[34]导入。经过构象搜索从催化剂的多个构象中获得能量最低的构象以及不同能量的构象用于生成加权平均构象描述符。计算中未考虑溶剂影响,停止优化的条件为Gaussian 的默认收敛标准。使用自研的程序对催化剂进行对齐,以提供更灵活和自动化的对齐方式。
在AI 模型设计和训练部分,所有的程序都是基于Pytorch 和Scikit-learn 实现,并将模型预测结果与已报道文献结果进行对比。鉴于数据规模较小,模型训练中使用了1 个NVIDIA GeForce GTX 1070,显存为8 GB。在数据预处理中,由于分子中原子数目不相同,故按列(特征)对数据进行统一长度和缩放处理,这有利于减少无效的数据、缩短描述符长度从而减少网络参数和提升效果。
3.3 主要结果
首先采用了基于加权平均占位的3D 描述符和分子级别的量子力学特征融合的模型对异恶唑存在下的Buchwald Hartwig 偶联反应进行筛选。在已有工作中,都使用传统机器学习算法来评判一个反应描述符的好坏,因此本文也做了类似的比较,如图6 所示。

图6 使用基于加权平均占位的3D描述符的6种方法在C-N偶联反应上的性能对比Fig.6 Performance comparison of six methods using 3D descriptors based on weighted average occupancy in C-N coupling reaction
在图6 所示的分析中,可以看到预测产率和观察产率的散点,其拟合出的直线应该满足如下规律:1)斜率为1。两种产率结果良好的拟合必然是拟合直线斜率为1。2)截距为0。截距能在一定程度上反映出预测结果相对于观察值之间的偏移。3)良好的散点疏密程度。真正有效的拟合必然是散点大部分聚集于拟合直线周围。
在随机划分(70/30,即训练和测试数据分别占70%和30%)数据集上,所提出的基于Boltzmann 分布的加权平均占位描述符可以使随机森林等机器学习方法得到较高的准确率,证明它对催化剂选择性的表达良好。其中,线性回归(图7(b))和SVM(图7(c))的截距大,斜率也不接近1,因而效果较差。K 近邻的斜率和截距都很好,但其中的散点大部分分散于距离拟合直线很远的区域,因而效果很差,自适应增强算法(Adaptive Boosting,AdaBoost)也存在同样的问题。表现好的是随机森林(图7(e))和本文的CNN 特征融合模型(图7(f)),而从散点的疏密程度来看,CNN 特征融合模型是最好的。图7 中使用了对映体过量(Enantiomeric Excess,ee)值指标来评估模型性能。
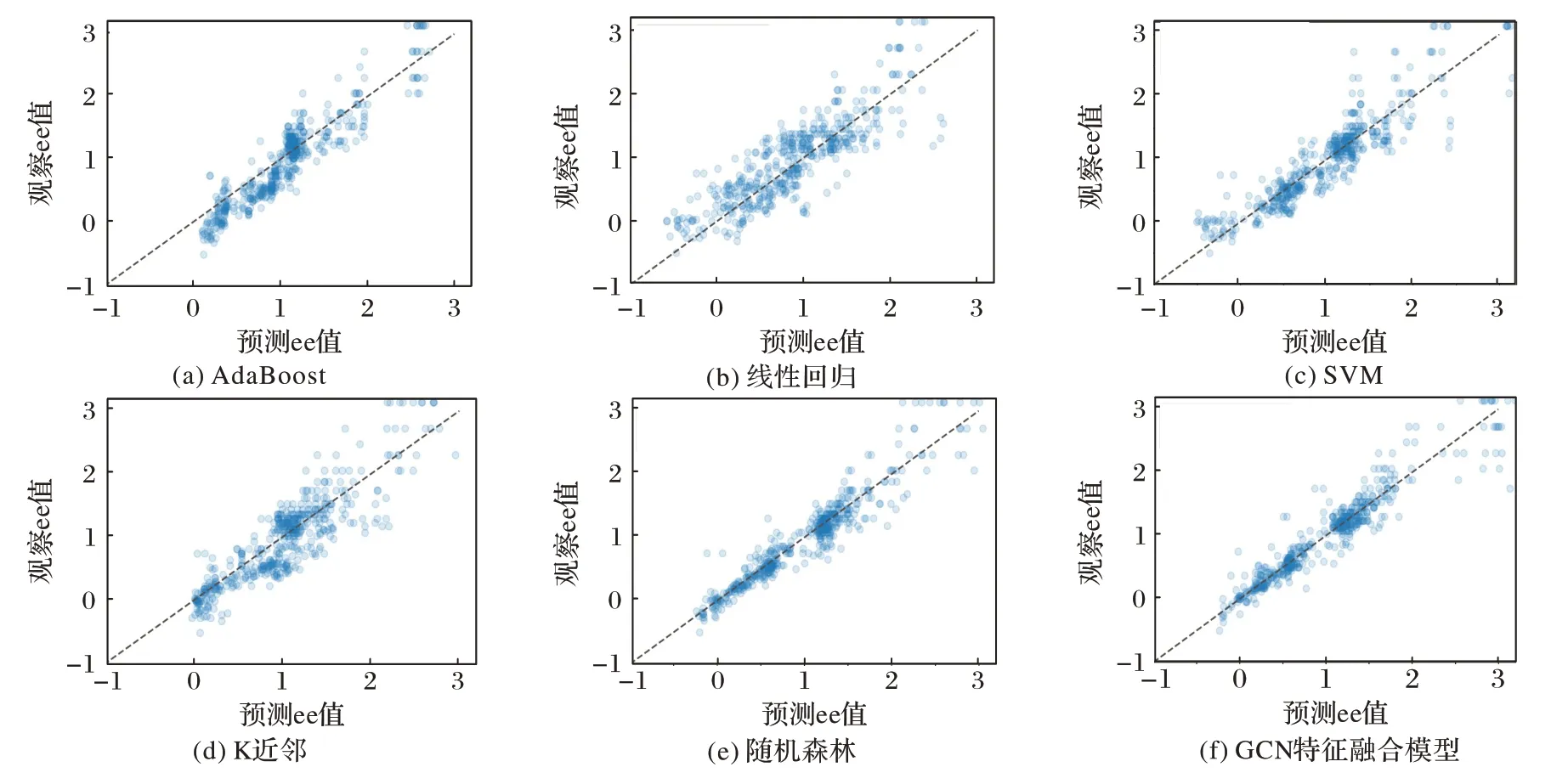
图7 使用基于量子力学特征与分子指纹的描述符的6种方法在N,S-缩醛反应上的性能对比Fig.7 Performance comparison of six methods using descriptors based on quantum mechanical features and molecular fingerprints in N,S-acetal formation
CNN 特征融合模型在随机划分数据上的R2比Ahneman[21]提高了3 个百分点,在additive test 2 数据集上比Schwaller[23]提高了1 个百分点,在additive test 4 数据集上比Ahneman[21]提高了2 个百分点(表3)。在4 个添加剂数据集上的平均性能比MFF[18]提高了8.5 个百分点,这说明了模型在添加剂水平不同的数据集中具有适用性。

表3 C-N偶联反应上的测试结果(R2)Tab.3 Prediction results(R2)on C-N coupling reaction
在手性磷酸催化的N,S-缩醛反应筛选中,使用的反应选择性评价指标是结合自由能差ΔΔG,实践中发现它比ee值的预测更有挑战性。ΔΔG的表达式为:
其中:R是理想气体常数,T是常温(取23℃)。
与前面的讨论类似,使用结合量子力学性质与分子指纹的描述符后,随机森林等传统机器学习方法表现出令人鼓舞的效果,这证明了描述符的有效性。在与已发表工作对比中,GCN 特征融合模型在多个数据集上的平均绝对误差(Mean Absolute Error,MAE)比MFF 降低1.2 个百分点,接近Zahrt 等[22]。在大多数数据集上取得了比基线方法更小的MAE(表4)。

表4 N,S-缩醛反应上的测试结果(MAE)Tab.4 Prediction results(MAE)on N,S-acetal formation
从已发表工作[18,21-22,35-36]可知,选择和构建分子描述符在有机反应预测中非常重要。实验结果表明,通过合适的特征融合模型将多种来源的描述符如量子力学性质、3D 空间性质、SMILES 和分子指纹等综合起来,在产率预测和选择性预测等筛选工作中都可以获得出色的预测性能,也是将深度学习中一些模型引入该领域的可选方案。
3.4 催化剂筛选结果
有效的催化剂的筛选可以大幅加快催化剂优化的进程。在手性磷酸催化的N,S-缩醛反应筛选中,通过对训练集之外的反应进行筛选,发现虽然训练集中包含大量的中、低反应选择性的催化剂和反应,高于95%的反应和催化剂很少,但GCN 特征融合模型却能把高选择性的催化剂大部分都预测出来,表现出令人鼓舞的外推能力。如图8 所示,GCN 特征融合模型可以筛选出有价值和高选择性的催化剂,且预测的ee 值与观察值非常接近。这种能力对于不对称催化的药 物合成反应的优化很有意义。

图8 使用GCN特征融合模型筛选出的一部分催化剂Fig.8 Some catalysts screened by GCN feature fusion model
本文通过分析与实验证明了使用深度学习方法融合多模态数据对药物合成反应进行虚拟筛选是可行的,并提出一种基于Boltzmann 分布的3D 描述符,它按构象能量计算权重从而将多个构象加权平均,来近似反应发生时构象的概率分布。针对单一模态分子特征表示的不足,提出一种融合3D描述符和DFT 分子性质的CNN 模型,用于4-甲基苯胺与芳基卤化物的C-N 偶联反应的产率预测;
同时还提出一种基于DFT 性质的图卷积网络,该网络将原子的QM 性质作为节点的输入特征,使用RDKit 产生的邻接矩阵来构建图结构,并利用分支结构将分子指纹特征融合进来。最后,在不对称N,S-缩醛反应上验证了这种基于图卷积的特征融合方法的有效性。因此,得出结论:使用量子力学性质构建的分子描述符,特别是3D 描述符是预测药物合成反应的有效特征表示,而且基于多模态融合的深度学习可以将它与SMILES 以及分子指纹等结合起来,协同发挥作用。