强化NADPH再生途径驱动手性β-羟基酯的高效合成
来源:优秀文章 发布时间:2023-01-18 点击:
杜惠君,高 晨,左振宇,李凌凌,杨忠华
(武汉科技大学化学与化工学院,湖北 武汉,430081)
β-羟基酯类手性醇广泛应用于医药合成和精细化学品领域[1],是他汀类药物、碳青霉烯类抗生素、L-肉毒碱等的关键手性砌块[2-3],可通过化学合成、外消旋体拆分和生物催化等方法制备[4],其中,全细胞生物催化不对称合成法因具有高度对映选择性、无胞内酶的分离提纯步骤且不需额外添加辅因子等优点而成为制备手性β-羟基酯的理想方法[5]。
目前,用于全细胞生物催化法不对称合成β-羟基酯的催化剂主要为微生物细胞[6],如Ma等[7]利用酵母细胞催化以4-氯乙酰乙酸乙酯(COBE)为模型底物的β-酮酯,不对称合成β-羟基酯的对映体过量值为92%。然而,利用野生型微生物细胞作为催化剂时缺乏足够的[H] 作为推动力[8],进而影响手性醇的合成效率。针对该问题,研究者通常将大肠杆菌(E.coli)作为表达宿主菌,利用代谢调控法构建羰基还原酶和还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)再生酶偶联体系,解决胞内辅酶NADPH的供给问题[9],从而改善微生物细胞的催化效率。据文献[10]报道,利用磷酸戊糖途径代谢强化NADPH再生能力可以提高β-羟基酯的合成效率,但该途径中存在CO2合成,致使碳源损失严重。而糖酵解(EMP)途径产生的副产物较少,理论上消耗1分子葡萄糖可产生2 分子的辅酶NADPH,故EMP可作为辅酶NADPH再生的主要途径[11]。大肠杆菌细胞中调控EMP再生NAD(P)H的甘油醛-3-磷酸脱氢酶(GAPDH)偏好辅酶为NAD+,不能再生NADPH,如果引入外源NADP+依赖性GAPDH(NADP+-GAPDH)取代NAD+依赖性GAPDH则可直接利用EMP途径再生NADPH[12]。另外,胞内NADP+含量不高也是造成胞内NADPH供给不足的原因,已有研究表明,NAD激酶是目前生物体内唯一能将NAD+磷酸化合成NADP+的酶,因此引入NAD激酶也可进一步促进NADPH的内源性合成[13]。本课题组[14]曾通过调控外源NADP+-GAPDH和过表达NAD激酶,大大提高了大肠杆菌胞内NADPH的内源性合成,(S)-苯乙醇的产率高达99.2%,对映体过量值(e.e.)超过99%,但该催化体系具有α-芳香酮类底物专一性,不适用于β-羟基酯的生产。而蜡样芽孢杆菌(Bacilluscereus)中的羰基还原酶对β-酮酯有较强的催化还原活性,且对辅酶NADPH亲和力较高,在中性环境下结构较稳定[15],基于此,本研究通过调控EMP途径中NADP+-GAPDH和过表达NAD激酶来强化NADPH的内源性合成,为β-酮酯的不对称还原过程提供足够的推动力,进而为手性β-羟基酯的高效合成提供可行性方案。
1.1 试剂、菌株和质粒
基因操作所用工具酶购自广州美基生物科技有限公司;
底物COBE购自上海麦克林生化科技有限公司;
乙酸乙酯、苯甲醛、葡萄糖、Na2HPO4、NaH2PO4等均为分析纯,购自于国药集团化学试剂有限公司。本研究所用菌株和质粒见表1,其中Ampr为氨苄青霉素,终浓度为100 μg/mL;
Kanr为卡那霉素,终浓度为50 μg/mL。所有细胞均用LB培养基进行培养,LB液体培养基由10 g胰蛋白胨、10 g NaCl、5 g酵母粉及1000 mL去离子水配制而成,此基础上另加入15 g琼脂粉配制LB固体培养基。

表1 菌株和质粒
1.2 重组质粒pETDueT-1-gapB-bcCR的构建
本研究利用双基因串联载体旨在构建羰基还原酶BcCR和NADP+-GAPDH再生NADPH偶联体系,其中BcCR为本课题组利用基因挖掘法获得的还原β-酮酯不对称合成β-羟基酯的高活性羰基还原酶,该酶的UniProt编号为Q819V6,GenBank核苷酸序列编号为AAP10771.1[15]。首先利用Primer Premier 5.0软件对照目的片段bcCR和载体质粒pETDueT-1的酶切位点信息设计引物,正向引物bcCR-F为GGGAATTCC↓ATATGATGTTAAAAGGGAAGGTAGCATT,其中C↓ATATG为Nde I的酶切位点,反向引物bcCR-R为CCC↓TCGAGCATTACCATACCGCCATCAAC,其中C↓TCGAG为Xho I的酶切位点。然后,以Bacilluscereus基因组DNA为模板,通过PCR扩增目的片段、双酶切、连接实验将bcCR导入质粒载体pETDueT-1和pETDueT-1-gapB中,并利用菌液PCR和双酶切鉴定目的基因的转化情况。图2所示为重组质粒pETDueT-1-gapB-bcCR构建原理。
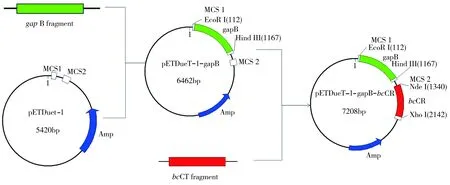
图1 重组质粒pETDueT-1-gapB-bcCR的构建
1.3 羰基还原酶偶联辅酶再生共表达体系的构建与表达
在代谢调控EMP途径中,源于Bacillussubtilis(strain 168)、编码基因为gapB的NADP+-GAPDH催化甘油醛-3-磷酸的不可逆氧化过程是大肠杆菌胞内NADPH再生的关键步骤,但其胞内氧化型辅酶NADP+供给不足,限制了NADPH再生。利用源于Escherichiacoli(strain K12 MG1655)、编码基因为yfjB的NAD激酶(NADK)可以使胞内NAD+磷酸化形成NADP+,从而为NADPH的再生提供足够的氧化型底物。故本研究将所构建的质粒pETDueT-1-bcCR和pETDueT-1-gapB-bcCR通过双质粒共表达技术电转至E.coliBL21(DE3)和E.coliBL21(DE3)/pET-28a-yfjB进行表达,获得重组大肠杆菌E.coliBL21(DE3)/pETDueT-1-bcCR、E.coliBL21(DE3)/pETDueT-1-gapB-bcCR、E.coliBL21(DE3)/pETDueT-1-bcCR&pET-28a-yfjB、E.coliBL21(DE3)/pETDueT-1-gapB -bcCR&pET-28a-yfjB,以E.coliBL21(DE3)/pETDueT-1&pET-28a为对照组。
利用0.4 mmol/L的IPTG对所构建的工程菌进行低温诱导表达后离心收集菌体,经浓度为0.05 mol/L、pH 为7.0的磷酸盐缓冲液(PBS)洗涤两次后,以0.1 g/mL的菌体浓度重悬细胞。采用超声破胞法提取粗酶液,利用聚丙烯酰胺凝胶电泳(SDS-PAGE)鉴定蛋白表达情况,同时,因BcCR、NADP+-GAPDH、NADK在催化过程中伴随NADPH的消耗或合成,且NADPH在340 nm处存在吸光值,故参考Qin等[15]所述方法测定BcCR的酶活,按Luo等[16]所述方法测定NADP+-GAPDH、NADK的酶活,并利用紫外可见分光光度法检测重组菌在24 h内的生长状况。
1.4 全细胞催化不对称还原COBE反应
全细胞催化不对称还原COBE的反应过程参照文献[16]。首先,使用pH为7.0、浓度为0.05 mol/L的PBS缓冲液以0.1 g/mL的菌浓度重悬大肠杆菌细胞。然后,取5 mL重悬液置于50 mL锥形瓶中,加入葡萄糖(1.5 g/L)和底物COBE(40 mmol/L),在37 ℃、180 r/min的条件下反应20 h。最后,利用乙酸乙酯对产物(R)-4-氯-3-羟基丁酸乙酯((R)-CHBE)和底物COBE进行萃取,以苯甲醛为内标物。
1.5 分析方法
1.5.1 重组蛋白酶的酶活检测方法
利用紫外/可见分光光度法[14-15]测定三种重组蛋白BcCR、NADP+-GAPDH、NADK的酶活力。在最适反应条件下,每分钟消耗或生成1 μmol NADPH所需要的酶量为一个酶活单位(U),即1 U=1 μmol/min。根据NADPH的标准吸光度曲线计算待测样品中NADPH的浓度CNADPH,计算公式为:
CNADPH=(A-0.016 55)/6.510 91
(1)
式中,A为吸光值。同时,以浓度为100 μg/mL的牛血清蛋白(BSA)为标准蛋白,利用考马斯亮蓝G-250染液法测定粗酶液中各蛋白的蛋白含量[15],进而分析3种重组酶的比活。蛋白含量Cprotein计算公式为:
Cprotein=(A+0.001 62)/0.008 97
(2)
蛋白酶比活的定义为:25 ℃下,单位质量的重组蛋白所具有的酶活力单位数。
1.5.2 COBE还原产物的定性和定量分析
借助Agilent 1100 HPLC型液相色谱仪,利用高效液相色谱法[17]确定目标产物的e.e.值,流动相为正己烷/异丙醇(体积比为85/15),流速为0.8 mL/min,柱温为25 ℃,检测波长为220 nm。e.e.值计算公式为:
(3)
式中,CR和CS分别是反应结束时产物(R)-CHBE和(S)-CHBE的浓度。
利用岛津GC-2010型气相色谱仪对COBE还原产物进行定量分析[15]。检测时以N2为载气,色谱柱为Rtx-Wax毛细管手性柱,流速为1.5 mL/min,分流比为30∶1。利用内标法计算目标手性醇产率Yield,计算公式为:
(4)
式中,Csub是反应初始时的底物浓度;
CP是反应结束时的产物浓度。
2.1 重组质粒pETDueT-1-gapB-bcCR的构建
利用双基因串联载体将外源性NADPH合成酶NADP+-GAPDH和羰基还原酶偶联表达,构建了重组质粒pETDueT-1-gapB-bcCR,重组质粒PCR鉴定结果如图2(a)所示,其中泳道M为Marker DL2000,泳道1为重组质粒pETDueT-1-bcCR的PCR鉴定结果,泳道2为重组质粒pETDueT-1-gapB-bcCR的PCR鉴定结果。由图2(a)可见,菌液PCR产物在750 bp处均存在单一目的条带,初步判断目的基因已成功导入。为了排除“假阳性”转化子的可能性,确定重组质粒是否构建成功,对重组质粒进行Nde I、Xho I双酶切,结果如图2(b)所示,其中泳道M为Marker DL5000,泳道1为pETDueT-1,泳道2为pETDueT-1-bcCR,泳道3为pETDueT-1-gapB-bcCR。从图2(b)中可以看出,对比泳道1中的空载质粒pETDueT-1,泳道3处获得了大小为6000 bp和750 bp左右的2个目的片段,分别与质粒pETDuet-1-gapB和目的基因bcCR的特征[11-12]相符,证实本研究已成功构建了质粒pETDueT-1-gapB-bcCR。

(a) PCR鉴定 (b)双酶切鉴定
2.2 羰基还原酶偶联辅酶再生体系的构建与表达
将所构建的工程菌进行诱导表达,利用超声破胞法提取目的蛋白,并通过SDS-PAGE凝胶电泳检测,结果如图3所示,其中泳道M为标准蛋白Marker,泳道1为源于E.coliBL21(DE3)/pETDuet-1&pET-28a粗酶液,泳道2为源于E.coliBL21(DE3)/pETDuet-1-bcCR粗酶液,泳道3为源于E.coliBL21(DE3)/pETDuet-1-gapB-bcCR粗酶液,泳道4为源于E.coliBL21(DE3)/pETDuet-1-bcCR&pET-28a-yfjB粗酶液;
泳道5为E.coliBL21(DE3)/pETDuet-1-gapB-bcCR&pET-28a-yfjB中的蛋白粗酶液。由图3可见,蛋白酶BcCR、NADP+-GAPDH和NADK的分子量分别为27、41、35 kDa,与文献[14-15]报道一致,与对照菌相比,上述3种蛋白酶均相应地成功表达出单一特异性条带,且表达效果良好,表明3者在重组大肠杆菌E.coliBL21(DE3)/pETDuet-1-gapB-bcCR&pET28a-yfjB中被高水平成功表达。

图3 SDS-PAGE检测结果
为了进一步探讨NADP+-GAPDH、NADK、BcCR共表达对大肠杆菌生长状况的影响,利用紫外/可见分光光度法测定了5种工程菌的生长情况,结果如图4所示。由图4可见,与对照菌E.coliBL21(DE3)/pETDueT-1&pET-28a相比,经IPTG诱导表达的其它4种工程菌生长速度远超过前者,表明利用双基因串联表达载体与双质粒共表达技术表达外源基因bcCR、gapB和yfjB可以大大改善宿主菌E.coliBL21(DE3)的生长状况,延缓重组菌进入生长稳定期的速度,且重组基因的表达对工程菌的影响无明显差异,这有助重组大肠杆菌用于全细胞催化不对称合成手性醇。

图4 重组大肠杆菌的生长
2.3 通过NADP+-GAPDH和NADK代谢调控胞内NADPH的合成
利用双基因串联载体将Bacillussubtilis(strain 168)中的NADP+-GAPDH在大肠杆菌中进行表达,引入糖酵解“旁路”以提高NADPH的内源性供给,相应检测结果如图5所示。由图5可见,相比E.coliBL21(DE3)/pETDueT-1-bcCR,在表达gapB基因的工程菌中,NADP+-GAPDH的比活提高了1.63倍,这与利用谷氨酸棒杆菌中的NADP+-GAPDH来强化NADPH合成能力的效果[18]相近,优于利用葡萄糖脱氢酶合成NADPH[19],这证实了调控EMP途径较PPP途径更有利于NADPH的内源性再生,而经共表达gapB和yfjB基因的工程菌E.coliBL21(DE3)/pETDueT-1-gapB-bcCR&pET-28a-yfjB胞内的NADP+-GAPDH比活提高了2.19倍,其NADPH再生能力分别是Martínez等[12]和Bommareddy等[20]所报道的仅利用NADP+-GAPDH强化NADPH供应时相应值的1.69和1.67倍,这表明NADK的过表达可以有效提高NADP+-GAPDH的转化效率,调控胞内NAD+代谢过程促进了NADP+的合成,为NADP+-GAPDH催化再生辅酶NADPH提供了足够的氧化型底物。

图5 BcCR、NADP+-GAPDH、NADK的比活测定
2.4 共表达体系在全细胞催化不对称还原COBE中的应用
选用COBE作为β-酮酯类化合物的模型底物,以(R)-CHBE和e.e.为评价指标测定共表达体系的转化效率,结果如图6所示。由图6可见,重组大肠杆菌E.coliBL21(DE3)/pETDueT-1-gapB-bcCR合成目标手性醇的产率是对照菌E.coliBL21(DE3)/pETDueT-1-bcCR相应值的1.38倍,其催化能力明显强于Ma等[7]所报道的野生型酵母细胞,是Ye等[19]所报道的不对称还原COBE转化效率的1.33倍。通过调控EMP强化NADPH内源性供给后,β-羟基酯的合成效率是利用葡萄糖脱氢酶(GDH)再生体系时相应值[21]的2.13倍,这进一步表明,与调控PPP途径中的GDH相比,调控EMP中的NADP+- GAPDH再生辅酶NADPH更能有效改善微生物细胞的催化效率。经NADP+- GAPDH和NADK共表达的工程菌E.coliBL21(DE3)/pETDuet-1-gapB-bcCR&pET28a-yfjB的催化效率相比对照菌相应值提高了1.69倍,(R)-CHBE产率高达98.18%,e.e.值超过99%,是朱清禾等[17]所获手性醇产率的3.27倍,而Zhang等[22]利用全细胞催化不对称合成(R)-CHBE的产率仅为89%,Houng等[23]全细胞催化双相体系还原COBE所获手性醇产率仅为74.5%,这表明在大肠杆菌中调控EMP和引入NAD激酶,可有效强化辅酶NADPH的内源性合成,提高β-酮酯类化合物的转化效率,有助于β-羟基酯类手性醇的高效合成。
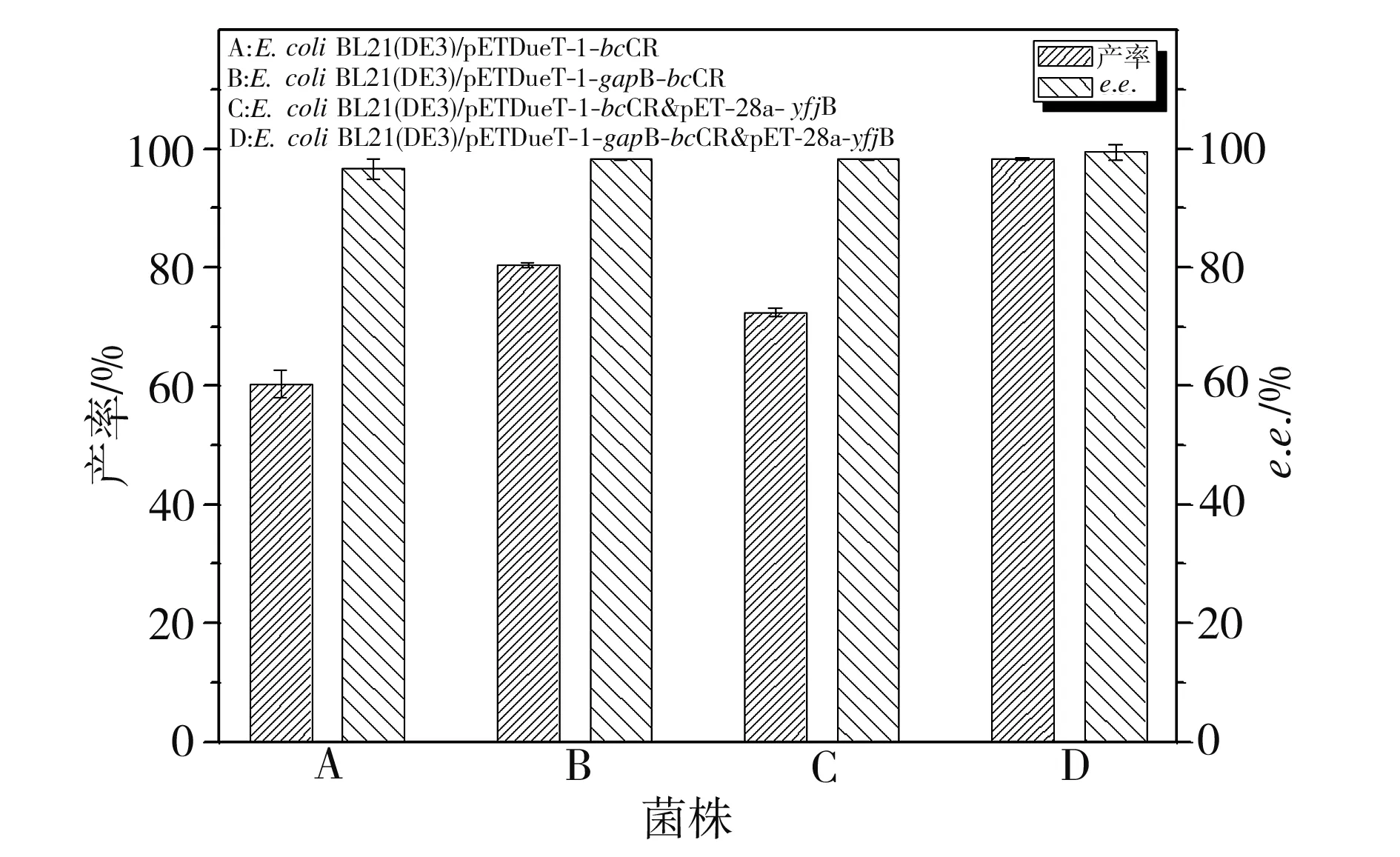
图6 产率与对映体过量值
本研究通过双基因串联表达载体和双质粒共表达体系构建羰基还原酶和辅酶NADPH再生偶联系统,定向改造大肠杆菌细胞内的EMP途径调控辅酶NADPH的内源性合成,构建了高效合成β-羟基酯类手性醇的工程菌E.coliBL21(DE3)/pETDuet-1-gapB-bcCR&pET28a-yfjB。该重组菌在不额外添加昂贵辅酶NADPH的前提下实现了β-羟基酯的高产、高立体选择性合成。以4-氯乙酰乙酸乙酯为模型底物,改造后的工程菌不对称合成(R)-4-氯-3-羟基丁酸乙酯的产率高达98.18%,对映体过量值超过99%,且其胞内辅酶NADPH合成能力相比E.coliBL21(DE3)/pETDueT-1-bcCR提高了2.19倍,这为β-羟基酯类手性醇合成提供了建设性和可行性方案。
猜你喜欢 辅酶羟基质粒 ——一道江苏高考题的奥秘解读和拓展">农杆菌转化法中的“Ti质粒转化载体系统”的简介——一道江苏高考题的奥秘解读和拓展中学生物学(2022年7期)2022-09-07国家药监局关于修订辅酶Q10注射剂说明书的公告(2022年第11号)中老年保健(2022年4期)2022-08-22全基因组测序后质粒的组装与鉴定研究进展*成都医学院学报(2022年4期)2022-08-19羟基磷石膏晶须制备及有机改性的研究建材发展导向(2021年7期)2021-07-16珊瑚羟基磷灰石表面改性的工艺昆明医科大学学报(2021年1期)2021-02-07开发新方法追踪植物病害的全球传播(2020.6.7 iPlants)三农资讯半月报(2020年11期)2020-06-21吃辅酶Q10有禁忌华声文萃(2019年4期)2019-09-10吃辅酶Q10有禁忌文萃报·周二版(2019年10期)2019-09-10小鼠转录因子STATl真核表达质粒的构建及生物学功能分析江苏农业学报(2019年1期)2019-09-10羟基化合物比较与例题剖析数理化学习·高一二版(2009年1期)2009-03-19推荐访问:羟基 高效 合成