新型SiAsP二维Janus材料电子结构和光吸收性质的第一性原理研究*
来源:优秀文章 发布时间:2022-12-10 点击:
岳子豪,张 会
(1. 沈阳大学 机械工程学院,沈阳 110044;
2. 沈阳大学 师范学院,沈阳 110044)
二维材料在垂直方向上存在着自由度的约束,电子在材料垂直方向上的移动被严格限制,因此具有很多区别于块体材料的优异特性[1-4]。2004年人们通过机械剥离的方法成功分离出石墨烯[5],由于具有超高的弹性模量、载流子迁移率和优异的光吸收能力等性质,石墨烯引起了人们对二维材料的广泛关注[6-7],此后黑磷烯[8-9]、硅烯[10]、过渡金属硫族化合物[11]等物性丰富的二维材料被相继探索。然而一些性能优异的二维材料在应用方面却会受限于自身特点,如石墨烯由于本征的零带隙无法应用于制备场效应晶体管[12-13],二维黑磷由于缺乏稳定性在空气和水中很快被氧化从而对性能造成影响[14-15],因此探索更多的新型二维材料仍然势在必行。
利用掺杂,使用同族或异族原子对已被探索材料中的原子进行替换是获取新型材料的常见方法[16-18],如Liang Dong等使用价电子同主族的Se原子对单层MoS2一侧的S原子进行替换,得到具有较强的压电效应的Janus结构MoSSe[1,19]; Yu等在单层MoSi2N4中的Si原子和Mo原子分别替换为同族Ge和W原子,得到具有优异的光催化分解水能力的Janus MoSiGeN4和Janus WSiGeN4[16]。
最近,一种二维IVA-VA化合物材料单层SiP2被报道[20],该材料是一种带隙大小为2.39 eV的间接半导体,其载流子迁移率达到3.27×104cm2/(V·s),光吸收性能优异,同时带隙可覆盖水的氧化还原电位,具有出色的光催化分解水能力。本工作使用第一性原理计算的方法对单层SiP2进行掺杂,使用同为第五主族的As原子,对SiP2中Si原子两侧的P原子进行替换,形成3种掺杂体系,得到SiAs2, α-SiAsP, β-SiAsP3种新型单层二维材料。并对这些材料的声子谱、晶体和电子能带结构与单层SiP2进行对比研究,验证出3种新型材料具有较高的结构稳定性,并在光学电子领域具有很好的应用前景。
本文研究使用基于密度泛函理论的第一性原理计算,采用Vienna Ab Initio Simulation Package (VASP)模拟软件包[21-22]进行。通过映射缀加波势来考虑电子-离子的相互作用。为了消除周期性计算导致的相邻结构之间的虚假相互作用,必要时在垂直方向上放置不小于1.5 nm的真空空间,使用Vaspkit代码对VASP计算出的数据进行后处理。
不同的函数可能会产生不同的密度泛函计算结果。由于晶体结构对计算细节的敏感性较低,因此最初使用具Perdew-Burke-Ernzerhof (PBE)泛函的广义梯度近似(GGA)[23]进行结构优化,并将能量截止值设置为500 eV。采用蒙克霍斯特包方案对布里渊区(BZ)[24]采取了15×7×1的K点网格采样,以确保原始六角形晶格计算的可靠性。晶格向量和原子坐标均松弛,每个原子的容差均小于0.1 eV/nm。正如在声比包中实现的,声子频率是通过基于有限位移方法引入位移计算的力常数得到的。声子计算采用包含54个原子的3×3×1超级细胞进行。利用Heyd-Scuseria-Ernzerhof (HSE06)杂化泛函[25]进一步优化了PBE优化后的结构,以获得更准确的结果。
2.1 晶体结构与稳定性
单层SiP2是一个由Si和P原子交互形成的弯曲蜂窝结构,形状与硅烯相似[26],晶体结构如图1所示。锯齿状链中的P1原子与Si原子相连,蜂窝结构中的每个Si/P原子与P/Si原子三配位,锯齿状链中的每个P原子连接一个Si和两个P原子,产生sp3杂化轨道。经过结构优化后,晶格常数a=0.345 nm,b=0.604 nm。Si-P键长为0.228 nm,P-P键长为0.227 nm,与已有研究结果数值完全一致[20],这表明本研究的计算结果是可靠的。

图1 (a)和(c)分别为单层SiP2晶体结构的俯视图与侧视图,(b)和(d)分别为单层SiP2中的蜂窝结构与锯齿状链结构Fig.1 (a), (c) the top and side view of monolayer SiP2 structure respectively,and (b), (d) honeycomb structure and zigzag chain structure in monolayer SiP2
本研究分别用As原子对SiP2中锯齿链状的P1原子、蜂窝结构中的P2原子以及P1和P2原子同时进行替换,形成3种新型二维结构,经过结构优化得到SiAs2, α-SiAsP, β-SiAsP3种新型单层二维材料,如图2所示。AS与P原子同属第五主族,最外层都有5个价电子,因此在对P原子替换后,As/Si原子之间的成键方式与P/Si原子相同,3种材料相对于SiP2在结构形状上未发生明显改变。由于As原子的半径大于P原子,3种材料的晶格常数以及共价键长相对于单层SiP2略微增大,具体数值如表1所示。其中单层SiAs2的晶格常数和共价键的键长增长最为明显,这是因为单层SiAs2是由As原子对单层SiP2两侧P原子同时替换得到的。本工作构造了两种单层SiAsP材料,α-SiAsP比β-SiAsP平均到每个原子的总能量低0.09 eV,表明前者更加稳定。图3中,单层SiAs2, α-SiAsP和β-SiAsP上下两侧静电势皆不相等,差值ΔΦ分为0.039, 0.567, 0.323 eV,因此上述材料在与平面垂直方向上具有非对称性,具有这种非对称性质的结构一般被称为Janus材料。
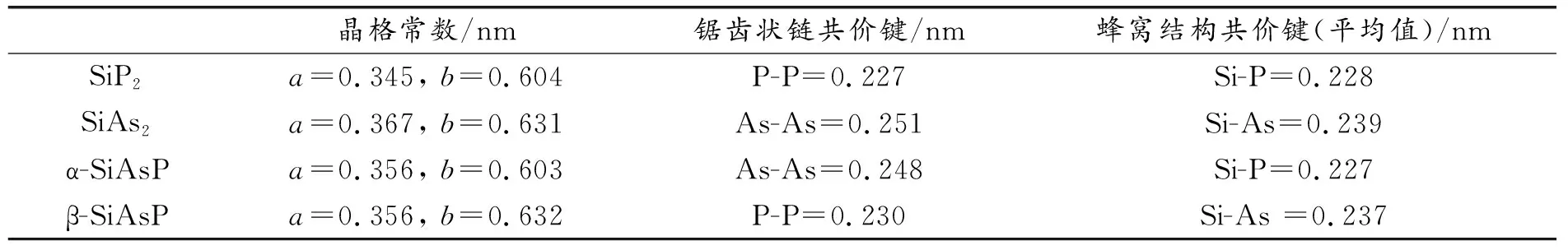
表1 4种材料的晶格常数与共价键长Table 1 Lattice constants and covalent bond lengths of four materials

图2 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP的晶体结构Fig.2 (a)-(c) The crystal structures of monolayer SiAs2, α-SiAsP and β-SiAsP

图3 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP的静电势Fig.3 (a)-(c) The electrostatic potentials of monolayer SiAs2, α-SiAsP and β-SiAsP
结构稳定性是二维材料得以实现的先决条件,声子谱计算是验证材料稳定性可靠方法。如图4所示。3种材料的声子谱中各有18条色散曲线(3条声学支,15条光学支),皆在零以上分布(G点附近出现部分虚频来自数值误差);
单层SiAs2, α-SiAsP, β-SiAsP的最高声子频率都位于Y点,分别可达12.79 THz (426 cm-1),15.32 THz (510 cm-1),14.38 THz (479 cm-1),略低于已被报道的单层SiP2的最高频率(529 cm-1)通过比较可知,3种材料中的最高频率与已报道的二维材料接近,如MoS2(473 cm-1)[11], CaP3(443 cm-1)[27], d-InP3(437 cm-1)[28]等,表明单层SiAs2, α-SiAsP, β-SiAsP 3种材料具有良好结构稳定性。

图4 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP的声子谱Fig.4 (a)-(c) The phonon dispersion curves of monolayer SiAs2, α-SiAsP and β-SiAsP
2.2 电子能带结构与性能
为了研究这3种材料的电子性能,利用PBE泛函计算了3种材料的电子能带结构,结果如图5所示。单层SiAs2, α-SiAsP和β-SiAsP在PBE水平上带隙值分别为1.53, 1.68, 1.03eV,表现出半导体的性质。3种材料的价带最大值(VBM)与导带最小值(CBM)分别位于不同的K点,(以图5(a)中SiAs2为例,其VBM位于G点,CBM位于Y点),因此可以判断这3种单层材料为间接带隙半导体。
众所周知,基于PBE函数计算出的电子带隙往往小于其实际值,为进一步探索3种单层材料的电子性质,利用HSE06杂化泛函计算了3种材料的电子结构和态密度。图5中单层SiAs2, α-SiAsP和β-SiAsP在HSE06水平上电子带隙分别为2.21, 2.43和1.76 eV。半导体的带隙大小与其光吸收能力相关,带隙在1.5~3 eV之间半导体可以满足高效太阳光利用的需要,并带隙大小为1.5 eV 附近的半导体光电转化效率可以达到最大值[29]。相比单层SiP2 (2.39 eV)[20],单层α-SiAsP,SiAs2的带隙值变化不大,而β-SiAsP的带隙值 (1.76 eV) 则与1.5 eV更为接近,说明β-SiAsP的光吸收效率可能优于原子替换前的单层SiP2。

图5 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP在PBE函数下的能带结构图;
(d)~(f)分别为单层SiAs2, α-SiAsP, β-SiAsP在HSE06函数下的能带结构图Fig.5 Electronic band structures of monolayer SiAs2, α-SiAsP and β-SiAsP from (a)-(c) PBE and (d)-(f) HSE06
此外,根据图6所示的总态密度(TODS)和部分原子态密度(PDOS)可知,单层SiAs2的VBM主要由锯齿状链中的As1原子和蜂窝结构中的As2原子贡献而来,CBM主要是由As1原子贡献的。单层α-SiAsP和β-SiAsP的VBM分别由As、P原子和P、Si原子贡献的,CBM分别由As, Si原子和As原子贡献的。

图6 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP在HSE06函数下态密度Fig.6 (a)-(c) The density of states of monolayer SiAs2, α-SiAsP, β-SiAsP from HSE06
2.3 光吸收性能
基于HSE06泛函,半导体的光吸收系数可以用频率相关的介电函数ε(ω) = ε1(ω)+iε2(ω)来表示,可通过α(ω)=√2(ω)[( ε12(ω)+ε22(ω))-ε1(ω)]来进行计算[30-31],其中ε12(ω)和ε22(ω)分别是介电函数的虚部和实部。计算结果得出,单层SiAs2, α-SiAsP, β-SiAsP在部分可见光(2.1~3.2 eV)和紫外范围内(>3.2 eV),光吸收系数能够达到105cm-1级别。其中单层β-SiAsP可见光的吸收范围比原子替换前的单层SiP2更广,这是原子替换后禁带变小的缘故。单层β-SiAsP的带隙(1.76 eV)最小,光吸收系数沿X和Y方向分别在1.9和2.2 eV出现峰值,能够同时吸收可见光和近红外光。因此,单层β-SiAsP在太阳能电池和光催化领域具有一定的应用潜力。
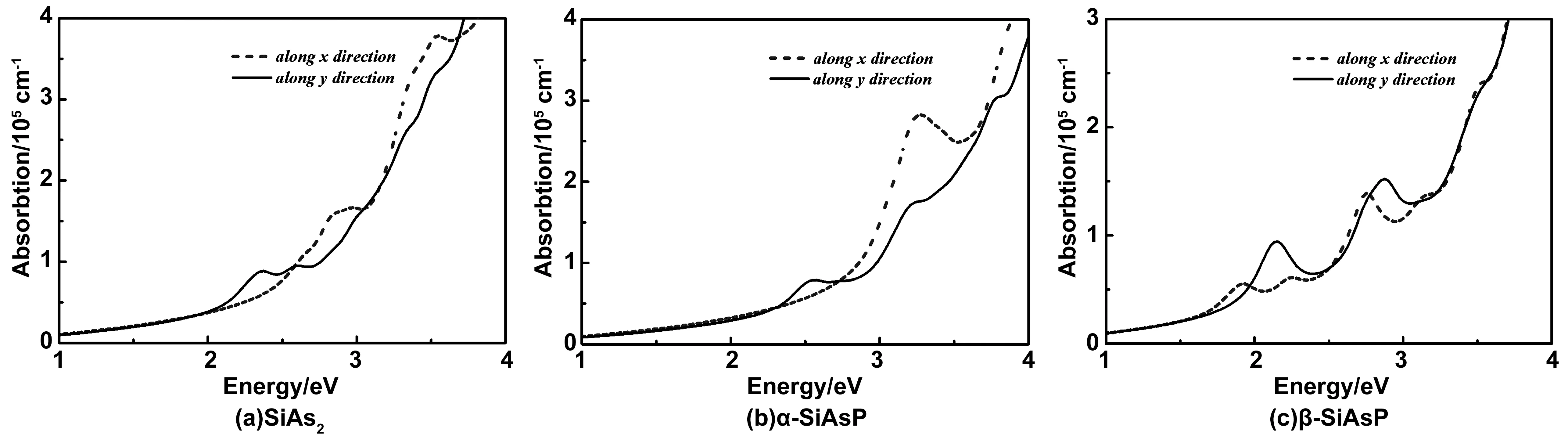
图7 (a)~(c)分别为单层SiAs2, α-SiAsP, β-SiAsP在HSE06函数下的吸收光谱Fig.7 (a)-(c) The optical absorption spectras of monolayer SiAs2, α-SiAsP and β-SiAsP from HSE06
基于第一性原理计算,预测了3种新型二维材料,单层SiAs2, α-SiAsP和β-SiAsP。由于是对单层SiP2进行原子替换得到的,3种材料的晶体结构差异较小,且都具有Janus结构。3种材料的声子色散曲线皆无虚频存在,这表明3种材料是稳定的,而同素异形体α-SiAsP相对于β-SiAsP能量更低,可判定α-SiAsP的稳定性高于β-SiAsP。通过计算3种材料的能带结构和态密度,得知3种材料为间接带隙半导体,在HSE06水平上带隙值分别为2.21,2.43和1.76 eV。通过计算3种材料的吸收光谱,验证出上述材料在大部分可见光范围和整个紫外区域的吸收系数可达105cm-1,其中β-SiAsP还可以在近红外区域有效地进行光吸收。结合3种材料的良好稳定性和半导体特性,预测出单层SiAs2, α-SiAsP和β-SiAsP 3种材料光学电子领域具有可观的应用前景。
猜你喜欢 能带晶格单层 二维四角TiC单层片上的析氢反应研究分子催化(2022年1期)2022-11-02含晶界单层过渡金属硫化物的压电效应研究南京航空航天大学学报(2022年4期)2022-08-30Lieb莫尔光子晶格及其光子学特性研究光子学报(2022年6期)2022-07-27二组元置换式面心立方固溶体晶格畸变的晶体学模拟沈阳大学学报(自然科学版)(2022年2期)2022-04-12张云熙作品选当代作家(2021年11期)2021-12-17吃东西时注意多小天使·聪聪画刊(2021年2期)2021-09-10电化学沉积制备高结晶度金箔中小企业管理与科技·上旬刊(2021年6期)2021-07-14汽车转向管柱吸能带变形研究和仿真优化汽车零部件(2020年10期)2020-11-09Life OR Death Decision汉语世界(The World of Chinese)(2019年6期)2019-09-10想你所想 不止于车汽车生活(2018年5期)2018-06-21推荐访问:吸收 性质 原理